Mukopolysacharidóza. Michalík, J., Zeman, J. a kol.
|
|
|
- Ladislava Dvořáková
- před 8 lety
- Počet zobrazení:
Transkript
1 Mukopolysacharidóza Michalík, J., Zeman, J. a kol.
2

3 Mukopolysacharidóza Jan Michalík, Jiří Zeman a kol.

4 Vydala: Společnost pro mukopolysacharidosu, 2010 Chaloupky Olomouc Tisk: Silueta Pardubice, s.r.o. Jazyková úprava: Zdenka Burešová, Ing. Grafická úprava: Jaroslav Pátek Kolektiv autorů: Jiří Zeman, prof. MUDr., DrSc. Jan Michalík, doc. Mgr. PaedDr., Ph.D., Lenka Vitnerová, MUDr. Pavel Ješina, MUDr., RNDr., Ph.D. Tomáš Honzík, MUDr., Ph.D. Martin Magner, MUDr. Marcela Staníková, Bc. Recenzovali: doc. MUDr. Jozef Hoza, CSc. doc. PhDr. Alena Petrová, CSc. ISBN: Tato publikace byla vydána za finanční podpory Úřadu vlády ČR a firem Genzyme Czech, s. r. o. a Shire HGT Czech Republic.

5 Obsah Úvod Lysosomální střádavá onemocnění Mukopolysacharidóza typ I (MPS I) Mukopolysacharidóza typ II (MPS II) Mukopolysacharidóza typ III (MPS III, Sanfilippo syndrom) Mukopolysacharidóza typ IV Mukopolysacharidóza typ VI Niemann-Pickova nemoc Veřejné zdravotní pojištění Zdravotnické prostředky Rodina a dítě nemocné mukopolysacharidózou Po sdělení diagnózy mluvte o svém údělu Reakce okolí a potřeba pomoci Odpovědnost rodičů Proměny nemoci a nutnost přizpůsobení Pečující matka a nemocné dítě Sourozenci nemocného dítěte Únava z dlouhodobé péče Nová situace v širší rodině Proměny času Mukopolysacharidóza jako dar? Závěr...110

6 Úvod Tato publikace je zaměřena na několik málo nemocí z rozsáhlé skupiny tzv. vzácných onemocnění. Jako vzácná onemocnění jsou označovány nemoci s velmi nízkým výskytem v populaci (obvykle pod 1 : 2000). V odborné literatuře již bylo popsáno více než 5800 různých typů vzácných onemocnění. I když některá z nich jsou supervzácná, dohromady je celkový výskyt nositelů vzácných onemocnění uváděn v rozmezí 3 až 4 % v populaci. V České republice to znamená každý rok až nových pacientů. Jednu skupinu supervzácných onemocnění tvoří lysosomální střádavá onemocnění. Lysosomální střádavá onemocnění patří do skupiny dědičných poruch metabolismu. V současné době je známo více než 40 lysosomálních onemocnění, z nichž některá se vyskytují skutečně mimořádně vzácně (výskyt v populaci od 1 : až k méně než 1 : ). Vyslovit slovo mukopolysacharidóza znamená dotknout se mimořádně závažných onemocnění. Mukopolysacharidózy - ve zkratce uváděné také jako MPS - jsou dědičně podmíněná onemocnění látkové výměny. Jsou to onemocnění, která jsou způsobena absencí některého životně důležitého enzymu. Tím dochází na některém stupni látkové výměny k narušení (bloku) přirozeně probíhajících proměn jednotlivých metabolitů. V organismu se potom hromadí produkty, které nemohou být dále odbourávány ani odstraněny a usazují se v buňkách životně důležitých orgánů, v játrech, v slezině, v srdci nebo v mozku. Napadené orgány omezují postupně svoje funkce a vyvolávají příznaky závažného onemocnění. Podle pořadí, v jakém byly objeveny nebo podle jmen lékařů a lékařek, kteří onemocnění popsali jako první, se MPS označují: MPS I (syndrom Pfaundlera-Hurlerové), později byla popsána i varianta tohoto typu s lehčím průběhem jako syndrom Scheie, MPS II (syndrom Hunter), typ MPS III (syndrom Sanfilippo), typ MPS IV (syndrom Morquio), typ MPS VI (syndrom Maroteaux Lamy) a velmi vzácný typ MPS VII (syndrom Sly). V České republice se ročně narodí několik dětí nemocných některou z forem mukopolysacharidózy či příbuzných onemocnění se střádáním, jako je např. nemoc Niemann-Pickova. Postupně se u nás zvyšuje informovanost dětských a praktických lékařů o těchto nemocech. Stále však - vzhledem k velmi nízkému výskytu nemá řada lékařů dostatečné zkušenosti s časnou diagnostikou onemocnění se střádáním. V případě chybných a pozdních diagnóz tak často dochází k nevratnému poškození zdraví či k řadě zbytečných vyšetření. U většiny nebo velké řady onemocnění se střádáním bylo zvykem dlouhá desetiletí psát přídomek nevyléčitelná. Vzhledem k nízkému výskytu byly výzkumné kapacity státních i soukromých laboratoří zaměřeny nejprve na onemocnění častější,

7 tj. nemoci, které postihují statisíce či miliony pacientů na celém světě. Teprve v poslední době dochází k objevům nových léků i pro pacienty se vzácnými poruchami. To se v plném rozsahu týká i mukopolysacharidóz. Charakteristickým znakem mukopolysacharidóz je mimořádná závažnost onemocnění, jeho těžký a progresivní průběh spojený s řadou somatických i funkčních změn organizmu nemocné osoby. Mukopolysacharidózy také zpravidla představují i mimořádnou zátěž nejen pro pacienta, ale i pro jeho rodinu. V naší publikaci přinášíme přehled nejnovějších poznatků o nejčastějších typech lysosomálních poruch, jejichž seznam budeme postupně rozšiřovat. Publikace je určena nejen zdravotnickým pracovníkům a rodičům nemocných dětí, ale i pedagogickým a sociálním pracovníkům, kteří přijdou do styku s dítětem s mukopolysacharidózou a jeho rodinou. Tomu odpovídá struktura knížky. V prvních kapitolách jsou uvedeny diagnostické, klinické a případně terapeutické aspekty jednotlivých druhů mukopolysacharidóz a Niemman-Pickovy choroby, které jsou určeny převážně lékařům. Rodiče, pedagogové, sociální pracovníci a další čtenáři mají možnost seznámit se s projevy onemocnění i se speciálními problémy dlouhodobé péče o nemocné dítě, které byly získány mnohaletými zkušenostmi desítek rodin s nemocným dítětem. Čtenáři jistě přivítají i kapitolu věnovanou možnostem (zajištění) prostředků zdravotnické techniky určených nemocným dětem. V publikaci jsou uvedeny údaje získané v rámci řešení grantového úkolu MZ ČR (č. MZ0 VFN 2005) a GA ČR (č. 406/09/ Kvalita života osob pečujících o člena rodiny s těžkým zdravotním postižením). Doc. Mgr. PaedDr. Jan Michalík, Ph.D předseda Společnosti pro mukopolysacharidózu Prof. MUDr. Jiří Zeman, DrSc přednosta Kliniky dětského a dorostového lékařství, VFN Praha

8 1. Lysosomální střádavá onemocnění Jiří Zeman Metabolismus (neboli látková výměna) je neustálý koloběh biochemických reakcí, přeměn a pochodů, při kterých se v buňkách tvoří chemické látky potřebné pro život a funkci buněk a tkání, ale současně se odbourávají i látky a odpadní produkty, které již nemají v těle uplatnění. Dědičné poruchy metabolismu vznikají při poruše funkce jednoho nebo více enzymů, které se podílejí na látkové výměně nebo změnách ve složení a množství strukturálních nebo transportních bílkovin. Lysosomální střádavá onemocnění představují širokou skupinu dědičných poruch metabolismu. Lysosomy jsou buněčné organely, ve kterých probíhá štěpení, odbourávání a odstraňování nebo přenos různých makromolekul (vysokomolekulárních látek). Lysosomální poruchy se klinicky projevují jako velice různorodá skupina více než 40 dědičně podmíněných onemocnění, při kterých má postižený pacient porušenu jednu nebo více lysosomálních funkcí. V důsledku lysosomální poruchy dochází u postižených osob k postupnému hromadění a střádání nerozložených a neodstranitelných velkých chemických molekul (makromolekul) uvnitř lysosomu. Zvětšené lysosomy mění tvar buněk, poškozují jejich funkci a pomalu, ale progresivně, vedou k typickým klinickým projevům onemocnění. Diagnóza lysosomálních onemocnění je obvykle založena na vyšetření aktivit jednotlivých enzymů v izolovaných leukocytech nebo kultivovaných fibroblastech, na histochemickém a elektron-optickém průkazu střádaného materiálu v biopsii z postižené tkáně a na molekulárně-biologickém vyšetření. Velice důležitá je včasná diagnostika, a to nejen pro účely genetického poradenství v postižené rodině, a dostupnost prenatální diagnostiky, ale i pro narůstající počet lysosomálních onemocnění, která jsou léčitelná transplantací hematopoietickými kmenovými buňkami (HSCT) nebo enzymovou substituční terapií (ERT, enzyme replacement therapy ). U pacientů s některými lysosomálními poruchami již byla úspěšně vyzkoušena i perorální léčba blokátory syntézy makromolekul a u řady dalších lysosomálních onemocnění se tato léčba zkouší. Genová terapie je zatím ve stádiu výzkumu. Mezi jednu z nejčastějších skupin lysosomálních onemocnění patří rozsáhlá skupina závažných onemocnění, které se označují jako mukopolysacharidózy. Podle typu enzymatického defektu i podle závažnosti klinického postižení se mukopolysacharidózy dělí na několik typů, jimž jsou věnovány následující části této publikace.

9 2. Mukopolysacharidóza typ I (MPS I) Pavel Ješina a Jiří Zeman Příčina onemocnění Mukopolysacharidóza I (MPS I) je dědičně podmíněné metabolické onemocnění ze skupiny lysosomálních střádavých onemocnění. MPS I je způsobena poruchou funkce enzymu alfa-l-iduronidázy, který se významně podílí na metabolismu a odbourávání glykosaminoglykanů (GAG). GAG jsou velké makromolekuly cukrů, které byly dříve označovány jako mukopolysacharidy. Podle střádání mukopolysacharidů v těle dostala tato skupina onemocnění i své označení - mukopolysacharidózy. Glykosaminoglykany se podílejí na stavbě pojivové tkáně a představují důležitou složku extracelulární matrix. Snížená nebo zcela chybějící aktivita enzymu alfa-l-iduronidázy způsobuje u pacientů pomalé ale progresivní hromadění a střádání GAG v mnoha tkáních s následnou poruchou jejich funkcí, které jsou v pokročilé fázi onemocnění již nevratné. Nejdříve se GAG hromadí jen v lysosomech, což vede pozvolna k jejich zvětšování, takže mohou být v buňkách viditelné optickým mikroskopem jako vakuoly, jak je tomu například v buňkách potních žlázek získaných při excizi kůže nebo v periferních lymfocytech v nátěru krevních buněk. Na biochemické úrovni jsou v moči vylučovány ve zvýšeném množství dermatansulfát a heparansulfát. Dědičnost a výskyt Dědičnost MPS I je autozomálně recesivní. To znamená, že k poruše funkce enzymu alfa- L-iduronidázy, který je kódován na dvou alelách (jedné alele paternální od otce a jedné alele maternální od matky), dochází pouze v případě, že oba rodiče jsou zdraví přenašeči pro MPS I a že jejich potomek získá od obou rodičů alelu, na které je přítomna mutace. Nemocný s MPS I tak má v genu pro MPS I mutace na obou alelách (je takzvaně homozygot pro danou mutaci), což vede k těžké poruše funkce alfa-l-iduronidázy s projevy onemocnění. Oba rodiče nemocného dítěte jsou klinicky zdrávi, oba mají mutaci pouze na jedné alele a jsou pro danou mutaci jen heterozygoti - přenašeči pro MPS I. V případě MPS I totiž pro dostatečnou funkci enzymu stačí přítomnost i pouze jedné nepostižené alely. MPS I postihuje rovnoměrně obě pohlaví, vyskytuje se celosvětově. Frekvence výskytu MPS I v populaci je přibližně 1: V genu pro MPS I již bylo popsáno více než 100 různých mutací. V České republice se nejčastěji vyskytují mutace označované jako W402X (cca u třetiny pacientů) a Q70X, které vedou k těžké poruše funkce enzymu a které spolu s mutací P533R představují více než polovinu mutovaných alel v kavkazské populaci. Zatím nebyla jednoznačně prokázána souvislost mezi genotypem a fenotypem onemocnění, což znamená, že neexistuje přímá souvislost mezi klinickým průběhem onemocnění a jeho prognózou a konkrétní mutací v genu pro MPS I. Jedinou výjimku

10 tvoří situace, kdy na obou alelách je přítomna tzv nulová mutace s úplnou ztrátou funkce, což se vždy projeví těžkým průběhem onemocnění. Klinické dělení MPS I na tři podtypy MPS I je pomalu progredující onemocnění, u kterého je popisována široká variabilita klinických příznaků s multisystémovým postižením, které může vyústit až k život ohrožujícím komplikacím. V minulosti se pacienti s MPS I podle závažnosti klinického průběhu onemocnění zařazovali do tří samostatných podskupin MPS I, ale škála různých projevů se u jednotlivých pacientů natolik překrývá, že řada autorů dnes hovoří o jednom onemocnění s velkou klinickou variabilitou. MPS I je tak chápána jako soubor klinických postižení, pohybujících se u pacientů od nejmírnějších projevů až po těžký fenotyp. Nejtěžší forma MPS I s nejzávažnější prognózou byla v roce 1919 pojmenována po doktorovi Pfaundlerovi a doktorce Hurlerové a označuje se jako MPS I (syndrom Pfaundler- Hurler, MPS I-H). Nejmírnější průběh onemocnění má MPS I-S (syndrom Scheie), pro který je typický pozdější začátek a mírnější klinické postižení. Tato forma MPS byl pospána americkým oftalmologem H. G. Scheie v roce Uprostřed klinického spektra těchto dvou forem onemocnění je syndrom Hurler-Scheie (MPS I HS). Jak jsme však již uvedli, klinické příznaky onemocnění představují spíše kontinuální spektrum od mírných projevů až po závažné. Navíc se závažnost jednotlivých příznaků v průběhu života mění, takže se v současnosti od toho dělení opouští. Pokud se ještě někde původní dělení MPS I na tři podtypy používá, tak to vychází spíše z tradice, než že by bylo možno použít přesných kritérií. Klinické projevy u pacientů s MPS I První příznaky těžší formy MPS I (původně MPS I-H), které zahrnují prohlubující se mentální retardaci a výrazné postižení pohybového aparátu, se začínají projevovat již v kojeneckém věku a postižené děti (pokud nejsou léčeny) umírají v důsledku progresivního neurologického postižení a kardiorespiračního selhání mezi rokem života. V kojeneckém věku je často přítomna makrocefalie, mírná hypotonie, hepatosplenomegalie, umbilikální a inguinální kýla. Později v batolecím věku se prohlubuje kraniofaciální dysmorfie s prominujícím čelem, sedlovitým nosem a makroglosií. Časté jsou infekty horních cest dýchacích, zvětšené adenoidní vegetace, porucha sluchu, zákal rohovky, hrudní kyfóza s protruzí sterna, gibbus v oblasti hrudní páteře, bederní hyperlordóza, omezená hybnost velkých i menších kloubů, drápovité postavení prstů na rukou, syndrom karpálního tunelu, porucha růstu, hypertrichóza a zpomalení a zástava psychomotorického vývoje. Ve školním věku dochází k postižení funkce srdečních chlopní a rozvoji kardiomyopatie. Závažnou komplikaci představuje rozvoj hydrocefalu a útlak krční míchy. Těžký průběh onemocnění se vyskytuje přibližně u poloviny ze všech dětí s MPS I.

11 U pacientů s přechodným fenotypem Hurler-Scheie (MPS I H-S) je průběh onemocnění mírnější i pomalejší a očekávaná doba života je delší (cca let). První příznaky onemocnění jsou obvykle popisovány až v batolecím nebo předškolním věku a deficit intelektu je zpočátku jen velmi malý nebo dokonce i žádný. Mezi první příznaky onemocnění, které jsou extrémně variabilní, mohou patřit hrubé rysy v obličeji, porucha růstu, zákal rohovky, ztuhlost kloubů, hluchota a postižení srdečních chlopní. Nejčastější příčinou úmrtí je srdeční selhání. Pro nejmírnější formu MPS I, Scheie syndrom (MPS I-S) je typický normální intelekt, mezi nejčastější obtíže patří degenerativní onemocnění kloubů, zákal rohovky a postižení srdečních chlopní. První příznaky onemocnění se objevují až kolem 5. roku a diagnóza MPS I je často stanovena až s velkým odstupem. Většina pacientů má normální délku života, největší riziko představují fatální kardiovaskulární a neurochirurgické komplikace. MPS I-S je vyskytuje přibližně jen u % pacientů s MPS I. Nepoměr vzhledem k vyššímu výskytu MPS I-H v populaci však může být způsoben i snadnější diagnostikou u pacientů s těžší formou onemocnění, zatímco řada pacientů s mírnou formou onemocnění může být dlouho vedena pod nesprávnou diagnózou. Dysmorfické změny a postižení muskuloskeletálního systému Změny v měkkých tkáních a kostní deformity jsou typickým znakem MPS I-H a vedou ke kraniofaciální dysmorfii, charakterizované zhrubnutím rysů v obličeji. U mírných forem onemocnění se tyto rysy projevují pomaleji nebo vůbec. U těžce postižených dětí jsou popisovány zpravidla makrocefalie s vysokým prominujícím čelem, makroglosie, sedlovitý nos s dopředu prominujícími nosními dírkami, gingivální hyperplázie. K dalším symptomům patří abnormální struktura vlasů, hypertrichóza, zašpičatělé zuby s vrozenou mezerou mezi předními řezáky (diastema), opožděné prořezávání zubů a kariézní chrup. Deformovaná sella tvaru J je typickým rentgeneologickým nálezem na lebce. Pacienti s MPS I mají disproporci těla s krátkým krkem s protruzí sterna a obvyklou hyperkyfózou v oblasti hrudní páteře až gibbem, které mohou být přítomny již od narození. Dospělí pacienti bývají většinou nižší postavy s abnormálním zakřivením páteře, často si stěžují na bolesti v bederní krajině. U všech pacientů s MPS I jsou pozorovány strukturní remodelace rostoucích kostí charakterizované jako dysostosis multiplex. To zahrnuje deformity dlouhých kostí, zašpičatělé metakarpální kůstky, dysplastické hlavice stehenních kostí, ovoidní těla obratlů se zobáčkovitým vytažením přední strany obratlů bederní páteře. Tvar žeber připomíná vesla zúžení u obratlového konce a naopak rozšíření u sternálního zakončení. Již v časném dětství bývá diagnostikována dysplázie kyčlí, která doprovází i deformity pánve. K akutním komplikacím patří míšní poranění v důsledku spondylolistézy a nestabilita atlanto-okcipitálního spojení způsobená dysplázií čepu 2. krčního obratle. Pacientům hrozí dislokace v kyčelním kloubu a typické je valgózní postavení kolenního kloubu. Progresivní artropatie, projevující se kontrakturami, může být patrna již ve věku 2 let, kdy jsou klouby postiženy symetricky, zpočátku bez známek

12 bolesti či zánětu. Jemná motorika ruky je zhoršena a může se vyvinout tzv. drápovitá ruka. Kromě toho se prakticky ve všech kloubech může vyskytnout ztuhlost, bolestivost a flekční kontraktury. Snižuje se vzdálenost, kterou je postižený pacient schopen ujít bez zastávky a lze pozorovat i chůzi po špičkách. Při progresivním postižení pohybového aparátu potřebují pacienti invalidní vozík. Gastrointestinální příznaky Mezi velmi časté příznaky patří inguinální a umbilikální hernie, které obvykle recidivují. Vznikají v prvních měsících života u nejtěžší formy MPS I-H a naopak u mírnějších forem jsou popisovány velmi vzácně a jsou spíše diskrétní. Jsou důsledkem kombinace oslabené pojivové tkáně stěny břišní a zvýšeného nitrobřišního tlaku díky hepatosplenomegalii. Hepatosplenomegalie se u pacientů s MPS I vyskytuje často, u mírné formy onemocnění může mít slezina i normální velikost. Zvětšení jater však nebývá spojeno s postižením jaterních funkcí. Velká hepatosplenomegalie může někdy vyústit v poruchu průchodnosti gastrointestinálního traktu se zácpou nebo bolestivými průjmy, dalšími projevy je zvracení nebo nepříjemné pocity předčasné sytosti. Kardiovaskulární systém Prakticky u všech pacientů s MPS I se mohou objevit patologické změny na srdečních chlopních, při kterých se progredující střádání GAG může projevit srdečními šelesty a později i stenózou nebo insuficiencí srdečních chlopní. Nejčastěji jsou postiženy aortální a mitrální chlopně s následnou dilatací srdce a kardiomyopatií s městnavým srdečním selháním. U některých pacientů s MPS I-H je kardiomyopatie přítomna již v 1. roce života. Právě kardiovaskulární obtíže vedou i u mírnějších forem MPS I k nejčastějším komplikacím. Respirační obtíže a ORL problematika Mezi časté postižení pacientů s MPS I-H patří obstrukce dýchacích cest, ke které dochází z důvodu zesílené mukózní membrány v nosohltanu, hrtanu a průdušnici. V této souvislosti je často popisována spánková apnoe-hypopnoe, která v kombinaci s hypoxickými podmínkami pravděpodobně zapříčiňuje postižení CNS. Hypoventilační syndrom s dyspnoí, vedoucí k atelektázám a častým plicním infektům, může nakonec způsobit plicní hypertenzi a cor pulmonale. Hypertrofie adenoidní tkáně je řešena i opakovanou adenotomií. Střádání GAG v orofaryngu, dysostóza ušních kůstek, poškození sluchového nervu a opakované mesotitidy způsobují částečnou nebo úplnou ztrátu sluchu. Senzori-neuronální hluchota může být zlepšena pomůckami (naslouchadla), ačkoli mentální úroveň pacienta je někdy limitující pro využití tohoto zařízení. 10

13 Neurologická projevy U pacientů s nejtěžší formou MPS I se ve druhé polovině prvního roku života zpomaluje vývoj kognitivních a motorických funkcí a celkový psychomotorický vývoj se v batolecím věku postupně zastavuje. V dalším průběhu neléčeného onemocnění dochází v batolecím a předškolním věku k regresi psychomotorického vývoje, což znamená, že postižené dítě zapomíná dovednosti, které se již dříve naučilo, Zhoršuje se řeč, klesá mentální kapacita dítěte a o něco později se zhoršuje i hrubá motorika a úroveň sociální i adaptivní chování. Za hlavní příčinu rychlé neurodegenerace se považuje stále větší hromadění GAG (především heparansulfátu) v nervových buňkách CNS. Častým nálezem u těžce postižených pacientů je komunikující hydrocefalus, projevující se rozšířením komorového systému CNS, jen vyjímečně dochází k rozestupu lebečních švů. V pokročilých stadiích onemocnění se může vyskytnout zvracení, bolesti hlavy a edém papily. Hromadění GAG v meningách způsobující jejich ztluštění včetně ligament se projevuje jako progresivní cervikální myelopatie, která může vyžadovat operační dekompresi míchy. Akutní míšní komprese může zapříčinit celé spektrum projevů MPS I. Zavedení shuntu může zmenšit problém, ale obvykle nemá vliv na onemocnění jako takové. Špatná funkce rukou je způsobena flekčními kontrakturami v kloubech a/nebo syndromem karpálního tunelu, ačkoli většina pacientů nevykazuje typické příznaky. Mezi další útlakové syndromy patří syndrom tarzálního tunelu a útlak míšního kořene. Oční postižení Mezi časté oční nálezy patří zákal rohovky, porucha zrakové ostrosti (popř. až slepota), glaukom a degenerace sítnice. Difúzní zákal rohovky spojený s přecitlivostí na světlo je příznakem u všech forem MPS I. U mírných případů byl zjištěn periferní zákal se zachováním dobré zrakové ostrosti. Může se vyskytnout glaukom, ale většinou je obtížně zjistitelný pro ztluštění rohovky, a tím komplikované možnosti změřit nitrooční tlak. U těžce postižených dětí může v důsledku dlouhodobého edému zrakového nervu dojít až k jeho atrofii. Degenerace pigmentového epitelu sítnice je doprovázena zhoršeným periferním viděním a noční slepotou. Diagnostika Diagnóza pacientů s MPS I závisí na klinickém podezření. Protože na počátku onemocnění nemusí být všechny projevy MPS I plně vyjádřeny, může být klinická diagnostika obtížná a často i opožděná, především u mírnější formy MPS I. Mezi chybné diagnózy může patřit diagnóza (juvenilní) revmatoidní artritidy, artrogrypóza, sklerodermie i některé degenerativní onemocnění a autoimunitní choroby. Po vyslovení klinické podezření na MPS, je prvním diagnostickým testem biochemické stanovení celkového množství a složení GAG v moči pomocí elektroforetické analýzy. 11

14 Toto vyšetření nerozliší s jistotou jednotlivé typy MPS a ani pozitivní nález ještě neznamená potvrzení diagnózy MPS. Pro definitivní stanovení diagnózy MPS I je nutné provést stanovení specifické enzymové aktivity alfa-l-iduronidázy v izolovaných leukocytech z periferní krve a/nebo v kultivovaných kožních fibroblastech. U pacientů s MPS I je enzymová aktivita obvykle snížená pod 2-5 % normy, ale zbytková aktivita nesouvisí se závažností klinických projevů. Pro účely genetického poradenství v rodině je vhodná diagnostika MPS I na molekulární úrovni pomocí mutační analýzy. Prenatální diagnostika MPS I je v ČR dostupná na biochemické i molekulární úrovni vyšetřením nativních nebo kultivovaných choriových klků, eventuálně kultivovaných amniocytů. Novorozenecký screening MPS I zatím není v České republice dostupný. Léčba Nezbytnou součástí léčby všech pacientů s MPS je sociální a symptomatická terapie. Původně byla paliativní a podpůrná léčba jedinou metodou pro postižené pacienty, která zahrnovala kromě sociální pomoci i rehabilitaci a dechovou rehabilitaci, podporu dýchání (tracheotomie a CPAP-continuous positive airways pressure ), transplantaci srdečních chlopní u srdečního selhávání, ortopedické korekce kostních abnormalit. V 80. letech minulého století byla poprvé realizována transplantace kostní dřeně. V současné době představuje transplantace hematopoietických kmenových buněk (HSCT) z kostní dřeně nejúčinnější léčbu a metodu volby u pacientů s nejtěžší formou MPS I, u kterých hrozí rozvoj postižení CNS. Podmínkou účinnosti transplantace (HCST) je nejen HLA identický dárce v rodině nebo v registru dárců ale i včasná diagnostika onemocnění (obvykle do 2 let věku), tedy ještě před rozvojem psychomotorické retardace. Samotná transplantace však má i určité riziko, například že transplantované buňky se tzv. nepřihojí. Metoda HCST není účinná u dětí s MPS I a významnějším postižením funkce CNS. Technologický vývoj v oblasti rekombinantních proteinů v posledních dvou dekádách umožnil vývoj tzv. enzyme replacement therapy (ERT, enzym substituční terapie). První úspěšná ERT byla provedena v devadesátých letech 20. století u pacientů s Gaucherovou nemocí, několik posledních let se používá i u pacientů s MPS I. Lékem volby je rekombinantní varianta enzymu alfa-l-iduronidázy, který je komerčně dostupný pod názvem Aldurazyme. Přípravek je aplikován intravenózně v kapací infuzi v týdenních intervalech. Jelikož enzym neproniká hemato-encefalickou bariérou, nemůže neovlivnit ani zabránit eventuálnímu rozvoji postižení CNS. Samostatná enzymoterapie je proto indikována u mírnějších forem MPS I bez významného postižení funkcí mozku. ERT se však úspěšně používá u malých dětí s těžkou formou MPS I před a v průběhu přípravy na transplantaci HCST. ERT je indikována i u některých pacientů s MPS I po transplantaci, pokud u nich 12

15 v důsledku vzniku chimerismu příliš poklesne aktivita vlastní alfa-l-iduronidázy, kterou lze monitorovat v izolovaných leukocytech periferní krve. Pravidelné intrathekální podávání ERT u pacientů s MPS I s postižením CNS je zatím ve fázi výzkumu. Studie vychází s předpokladu, že přímé podání enzymu do mozkomíšního moku může překlenout současný problém hemato-encefalické bariéry. Ve fázi výzkumu je i perorální substrát-inhibiční terapie MPS I pomocí malé molekuly. Také genová terapie MPS I je dosud jen ve fázi výzkumu na úrovni experimentálních zvířat. 13

16 3. Mukopolysacharidóza typ II (MPS II) Martin Magner a Jiří Zeman Mukopolysacharidóza typ II (MPS II, Hunterova nemoc) je závažné metabolické onemocnění, které patří do skupiny lysosomálních onemocnění se střádáním. Příčiny onemocněni Mukopolysacharidóza typ II je dědičně podmíněné onemocnění, která vzniká při poruše funkce lysosomálního enzymu iduronátsulfatázy. Iduronátsulfatáza je podobně jako všechny ostatní bílkoviny v těle kódována genetickou informací uloženou v genech. Chyba v genetickém zápisu se nazývá mutace. Na základě geneticky chybné informace si organismus si vyrábí enzym, jehož aktivita je výrazně snížená (tzv. zbytková) nebo dokonce zcela nulová, takže enzym nemůže účinně vykonávat svou biologickou funkci. Iduronátsulfatáza se podílí na metabolismu a odbourávání velkých molekul (makromolekul), které se nazývají mukopolysacharidy neboli glykosaminoglykany (zkráceně GAG), které představují důležitou složku mezibuněčné hmoty, (tzv. extracelulární matrix). GAG se ve větším množství vyskytují především v pojivové tkáni. Díky svým chemickým vlastnostem výrazně určují mechanické vlastnosti tkání, jejich pevnost i elasticitu. GAG se pravděpodobně účastní i adheze buněk, jejich růstu a diferenciace i mezibuněčných vztahů a interakcí. Během života se mezibuněčná hmota neustále tvoří i odbourává, což vede k velkému dennímu obratu metabolismu GAG. Štěpení chemicky složitých GAG probíhá uvnitř buněk v buněčných organelách, které se nazývají lysosomy, postupně a vyžaduje přítomnost celé řady po sobě fungujících enzymů. Jedním z těchto enzymů je iduronátsulfatáza. Nerozložené GAG se v lysosomech postupně hromadí, zvětšují jejich velikost a postupně vyplní celou buňku. Klinické projevy onemocnění vznikají v situaci, kdy postižené buňky již ztrácejí schopnost svých základních biologických funkcí Výskyt Mukopolysacharidóza typ II patří mezi vzácná nemocnění. Vyskytuje se přibližně u jednoho chlapce ze 120 až 160 tisíc. U dívek se MPS II vyskytuje zcela ojediněle, dosud bylo popsáno méně než 10 případů. Dědičnost Dědičnost mukopolysacharidózy typ II je na rozdíl od všech ostatních typů MPS, které mají dědičnost autosomálně recesivní, je gonosomálně recesivní a je vázána na pohlavní chromozom X. V genu pro iduronátsulfatázu bylo popsaných více než 300 různých mutací. Nejčastější jsou mutace bodové (asi 79 %), menší část mutací (cca 19 %) tvoří dalece, což je absence větší části genu. Přibližně u jednoho procenta nemocných chlapců se nepodaří genetický podklad onemocněné (mutaci) rozpoznat. 14

17 Zatímco ženy a dívky mají 2 chromozomy X, které získávají po jednom z vajíčka matky a spermie otce, mají muži a chlapci jen jeden chromozom X (lokus Xq28.1), který dostali z vajíčka matky. Otec svým synům chromozom X nepředává. Znamená to, že při normální funkci a počtu chromozomů X mohou mukopolysacharidózou typ II onemocnět pouze chlapci, zatímco děvčata s jedním postiženým chromozomem X jsou klinicky zdráva, ale mohou v dospělosti postižený chromozom X předat svým dětem. Pokud postižený chromozom X předají synovi, chlapec onemocnění mukopolysacharidózou typ II. Pokud jej předají své dceři, bude dívka klinicky zdravá, ale bude přenašečka jako její matka. Dívka může onemocnět jen velice vzácně. Historie Mukopolysacharidózy provázejí lidstvo od nepaměti. Různá vyobrazení pacientů s výrazem nápadně připomínajícím mukopolysacharidózu lze vysledovat do středověku. Kanadskou rodinu chlapce s mukopolysacharidózou typ II popsal ve svém románu severoamerický spisovatel Irving Washington ( ). To, že se jednalo právě o tuto nemoc, lze doložit i na základě klasického rodokmenu postižených členů rodiny. Onemocnění dostalo své jméno podle skotského lékaře Charlese A. Huntera, který v odborném časopise v práci z roku 1917 poprvé popsal typické klinické příznaky onemocnění u dvou postižených chlapců. Klinické příznaky onemocnění Protože porušená funkce iduronátsulfatázy postihuje celý systém štěpení GAG v těle, mohou se klinické projevy u jednotlivých typů mukopolysacharidóz částečně překrývat. Navíc při různé zbytkové aktivitě enzymu je míra postižení u MPS II velice variabilní. Nejtěžší typ MPS II může probíhat jako těžké progresivní život ohrožující onemocnění s manifestací v časném kojeneckém nebo batolecím věku (typ A), naopak mírné postižení se obvykle projevuje později, a má lepší prognózu (typ B). Mezi oběma typy onemocnění však neexistuje přesná hranice a jednotlivé klinické projevy se mohou překrývat. To vedlo některé autory k názoru, že je nutno tuto uměle vytvořenou klasifikaci považovat za čistě pomocnou. Klinické projevy onemocnění jsou málokdy patrné již v novorozeneckém věku a většina postižených chlapců odchází z porodnice jako ostatní zdravé děti. První příznaky jsou vesměs nespecifické. Patří mezi ně pupeční nebo tříselná kýla, sedlovitý nos, větší obvod hlavy, opakované záněty horních cest dýchacích, časté záněty středouší či omezená ztuhlost kloubů, například loketních. U chlapců s těžším typem MPS II se ke konci kojeneckého věku a na začátku batolícího věku objevuje hepatomegalie, změny v obličeji, zpomaluje se růst a opožďuje se vývoj v oblasti hrubé motoriky. Zvětšuje se jazyk, vývoj řeči je často opožděn. Onemocnění obyčejně postupuje pomalu, takže rodiče si probíhajících změn u dítěte nemusí hned všimnout. Trvá několik let, než se nahromadění 15

18 (střádání) mukopolysacharidů projeví tak charakteristickými známkami, které rozpozná i laik. Střádání v játrech a slezině vede k progredující hepatosplenomegalii. Postižení skeletu se projeví poruchou růstu i změnami ve tvaru a struktuře kostí s následnou protruzí sterna, hyperlordózou bederní páteře a hrbem v oblasti hrudní páteře. Postupně se mění i fyziognomie obličeje se záklonem hlavy, takže postižení chlapci si jsou navzájem velice podobní, jako kdyby všichni pocházeli z jedné rodiny. V této fázi onemocnění, obvykle mezi rokem života již lze u chlapců s těžším typem MPS II klinickou diagnózu snadno stanovit, i když zkušený klinik může na MPS II pomýšlet dříve. Postava a vzhled Chlapci s MPS II mají charakteristický vzhled i postavu s mírným předklonem trupu a dopředu vystupujícím břichem, což způsobuje nejen zvětšení jater a sleziny, ale i snížená síla břišního svalstva. Pro zvýšený nitrobřišní tlak a nedostatečnou pevnost a elasticitu pojiva se často vytváří pupeční nebo tříselná kýla. Krk bývá zkrácen, hrudník je často soudkovitý. Fyziologické zakřivení bederní páteře je zvýrazněno (bederní hyperlordóza). Různě vyjádřena je hrudní kyfóza, která může imponovat až jako hrb. Dolní i horní končetiny jsou mírně pokrčené pro kontraktury v loketních a kolenních kloubech. Ruce jsou drobné a prsty na rukou jsou zkrácené, jako kdyby ztluštělé a nelze je pro kontraktury narovnat. Růst je méně postižen než u pacientů s Hurlerovým syndromem, variabilita je však i tady veliká. Ve věku 10 let může porucha růstu u postižených chlapců dosáhnout deficitu cm. Obvod hlavy bývá v prvních dvou letech života zvětšený (tzv. makrocefalie). Nápadné je výrazné a husté obočí, které může ve střední čáře splývat. Vlasy jsou husté, vlasová hranice bývá položena nízko. Kořen nosu je vpadlý, nostrily obrácené směrem dopředu (antevertované), rty bývají ztluštělé. Lícní kosti čnějí dopředu a čelisti jsou předsunuté. Mluví se o tzv. hrubých rysech obličeje. Jazyk je zvětšený (makroglossie), mohou být přítomny tvarové změny zubů. Hlas je jako hluboký a chraplavý. Projevem střádání v kůži a podkoží jsou drobné i větší podkožní uzlíčky, které u některých chlapců tvoří tzv. hrbolatou kůži, zejména na ramenou a zádech, ale vyskytují se i na stehnech. Mohou se objevit a později vymizet nebo se znovu vrátit. Postižení srdce a plic Mukopolysacharidy se ve zvýšené míře ukládají i na srdečních chlopních, jejichž postižení se nejdříve projevuje srdečním šelestem a až později chlopenními vadami, které následně vedou až k celkovému postižení srdce s projevy srdečního selhávání. Primární hypertrofická kardiomyopatie se u mukopolysacharidózy typ II prakticky nevyskytuje. Postižení plic vede ke snížení plicních objemů a spolu s postižením srdce vede k nižší tolerancí fyzické námahy. Časté jsou chronická rýma, zvětšení nosních i krčních mandlí a obstruktivní spánková apnoe. Některé děti mívají i opakované zápaly plic. 16

19 17
20 Štěpánův úsměv na jednom setkání Společnosti Štěpán s maminkou 18

Periodické syndromy asociované s kryopyrinem (CAPS)
") www.printo.it/pediatric-rheumatology/cz/intro Periodické syndromy asociované s kryopyrinem (CAPS) Verze č 2016 1. CO JE CAPS 1.1 Co je to? Periodické syndromy asociované s kryopyrinem (CAPS) nebo také
www.printo.it/pediatric-rheumatology/cz/intro Periodické syndromy asociované s kryopyrinem (CAPS) Verze č 2016 1. CO JE CAPS 1.1 Co je to? Periodické syndromy asociované s kryopyrinem (CAPS) nebo také
VNL. Onemocnění bílé krevní řady
 VNL Onemocnění bílé krevní řady Změny leukocytů V počtu leukocytů Ve vzájemném zastoupení morfologických typů leukocytů Ve funkci leukocytů Reaktivní změny leukocytů Leukocytóza: při bakteriální infekci
VNL Onemocnění bílé krevní řady Změny leukocytů V počtu leukocytů Ve vzájemném zastoupení morfologických typů leukocytů Ve funkci leukocytů Reaktivní změny leukocytů Leukocytóza: při bakteriální infekci
Genetické aspekty vrozených vad metabolismu
 Genetické aspekty vrozených vad metabolismu Doc. MUDr. Alena Šantavá, CSc. Ústav lékařské genetiky a fetální medicíny FN a LF UP Olomouc Johann Gregor Mendel (1822-1884) Sir Archibald Garrod britský pediatr
Genetické aspekty vrozených vad metabolismu Doc. MUDr. Alena Šantavá, CSc. Ústav lékařské genetiky a fetální medicíny FN a LF UP Olomouc Johann Gregor Mendel (1822-1884) Sir Archibald Garrod britský pediatr
ZÁKLADY FUNKČNÍ ANATOMIE
 OBSAH Úvod do studia 11 1 Základní jednotky živé hmoty 13 1.1 Lékařské vědy 13 1.2 Buňka - buněčné organely 18 1.2.1 Biomembrány 20 1.2.2 Vláknité a hrudkovité struktury 21 1.2.3 Buněčná membrána 22 1.2.4
OBSAH Úvod do studia 11 1 Základní jednotky živé hmoty 13 1.1 Lékařské vědy 13 1.2 Buňka - buněčné organely 18 1.2.1 Biomembrány 20 1.2.2 Vláknité a hrudkovité struktury 21 1.2.3 Buněčná membrána 22 1.2.4
Paliativní péče u nervosvalových onemocnění v dětském věku
 Paliativní péče u nervosvalových onemocnění v dětském věku Jana Haberlová Neuromuskulární centrum FN Motol Klinika dětské neurologie 2.LF UK a FN Motol Obsah přednášky Charakteristika nervosvalových onemocnění
Paliativní péče u nervosvalových onemocnění v dětském věku Jana Haberlová Neuromuskulární centrum FN Motol Klinika dětské neurologie 2.LF UK a FN Motol Obsah přednášky Charakteristika nervosvalových onemocnění
Cévní mozková příhoda. Petr Včelák
 Cévní mozková příhoda Petr Včelák 12. 2. 2015 Obsah 1 Cévní mozková příhoda... 1 1.1 Příčiny mrtvice... 1 1.2 Projevy CMP... 1 1.3 Případy mrtvice... 1 1.3.1 Česko... 1 1.4 Diagnóza a léčba... 2 1.5 Test
Cévní mozková příhoda Petr Včelák 12. 2. 2015 Obsah 1 Cévní mozková příhoda... 1 1.1 Příčiny mrtvice... 1 1.2 Projevy CMP... 1 1.3 Případy mrtvice... 1 1.3.1 Česko... 1 1.4 Diagnóza a léčba... 2 1.5 Test
GLAUKOM. Autor: Kateřina Marešová. Školitel: MUDr. Klára Marešová, Ph.D., FEBO. Výskyt
 GLAUKOM Autor: Kateřina Marešová Školitel: MUDr. Klára Marešová, Ph.D., FEBO Výskyt Glaukom, laicky označovaný jako zelený zákal, je skupina očních chorob, které jsou charakterizovány změnami zrakového
GLAUKOM Autor: Kateřina Marešová Školitel: MUDr. Klára Marešová, Ph.D., FEBO Výskyt Glaukom, laicky označovaný jako zelený zákal, je skupina očních chorob, které jsou charakterizovány změnami zrakového
Deficit antagonisty IL-1 receptoru (DIRA)
") www.printo.it/pediatric-rheumatology/cz/intro Deficit antagonisty IL-1 receptoru (DIRA) Verze č 2016 1. CO JE DIRA? 1.1 O co se jedná? Deficit antagonisty IL-1Receptoru (DIRA) je vzácné vrozené onemocnění.
www.printo.it/pediatric-rheumatology/cz/intro Deficit antagonisty IL-1 receptoru (DIRA) Verze č 2016 1. CO JE DIRA? 1.1 O co se jedná? Deficit antagonisty IL-1Receptoru (DIRA) je vzácné vrozené onemocnění.
GENETIKA. Dědičnost a pohlaví
 GENETIKA Dědičnost a pohlaví Chromozómové určení pohlaví Dvoudomé rostliny a gonochoristé (živočichové odděleného pohlaví) mají pohlaví určeno dědičně chromozómovou výbavou jedince = dvojicí pohlavních
GENETIKA Dědičnost a pohlaví Chromozómové určení pohlaví Dvoudomé rostliny a gonochoristé (živočichové odděleného pohlaví) mají pohlaví určeno dědičně chromozómovou výbavou jedince = dvojicí pohlavních
Diagnostika a příznaky mnohočetného myelomu
 Diagnostika a příznaky mnohočetného myelomu J.Minařík, V.Ščudla Mnohočetný myelom Nekontrolované zmnožení nádorově změněných plasmatických buněk v kostní dřeni Mnohočetný = obvykle více oblastí kostní
Diagnostika a příznaky mnohočetného myelomu J.Minařík, V.Ščudla Mnohočetný myelom Nekontrolované zmnožení nádorově změněných plasmatických buněk v kostní dřeni Mnohočetný = obvykle více oblastí kostní
Syndrom fragilního X chromosomu (syndrom Martinův-Bellové) Antonín Bahelka, Tereza Bartošková, Josef Zemek, Patrik Gogol
 Antonín Bahelka, Tereza Bartošková, Josef Zemek, Patrik Gogol") Syndrom fragilního X chromosomu (syndrom Martinův-Bellové) Antonín Bahelka, Tereza Bartošková, Josef Zemek, Patrik Gogol 20.5.2015 Popis klinických příznaků, možnosti léčby Muži: střední až těžká mentální
Syndrom fragilního X chromosomu (syndrom Martinův-Bellové) Antonín Bahelka, Tereza Bartošková, Josef Zemek, Patrik Gogol 20.5.2015 Popis klinických příznaků, možnosti léčby Muži: střední až těžká mentální
Registrační číslo projektu: CZ.1.07/1.5.00/34.0649. Základy genetiky - geneticky podmíněné nemoci
 Výukový materiál zpracován v rámci projektu EU peníze školám Název školy: Střední zdravotnická škola a Obchodní akademie, Rumburk, příspěvková organizace Registrační číslo projektu: CZ.1.07/1.5.00/34.0649
Výukový materiál zpracován v rámci projektu EU peníze školám Název školy: Střední zdravotnická škola a Obchodní akademie, Rumburk, příspěvková organizace Registrační číslo projektu: CZ.1.07/1.5.00/34.0649
Hemofilie. Alena Štambachová, Jitka Šlechtová hematologický úsek ÚKBH FN v Plzni
 Hemofilie Alena Štambachová, Jitka Šlechtová hematologický úsek ÚKBH FN v Plzni Definice hemofilie Nevyléčitelná vrozená krvácivá choroba s nedostatkem plazmatických faktorů FVIII hemofile A FIX hemofile
Hemofilie Alena Štambachová, Jitka Šlechtová hematologický úsek ÚKBH FN v Plzni Definice hemofilie Nevyléčitelná vrozená krvácivá choroba s nedostatkem plazmatických faktorů FVIII hemofile A FIX hemofile
Pacient s hemofilií. Radomíra Hrdličková
 Pacient s hemofilií Radomíra Hrdličková Hemofilie Vrozené krvácivé onemocnění (Genetická porucha sekundární hemostázy) Hemofilie A - deficit koagulačního faktoru VIII Hemofilie B - deficit koagulačního
Pacient s hemofilií Radomíra Hrdličková Hemofilie Vrozené krvácivé onemocnění (Genetická porucha sekundární hemostázy) Hemofilie A - deficit koagulačního faktoru VIII Hemofilie B - deficit koagulačního
 http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
Stanovení pojistného plnění z pojištění trvalých následků úrazu
 trvalé následky 1 / 7 úrazu. Stanovení pojistného plnění z pojištění trvalých následků úrazu Co musí trvalý následek splňovat Pojistné krytí se vztahuje výhradně na trvalé následky úrazu, nikoli nemoci.
trvalé následky 1 / 7 úrazu. Stanovení pojistného plnění z pojištění trvalých následků úrazu Co musí trvalý následek splňovat Pojistné krytí se vztahuje výhradně na trvalé následky úrazu, nikoli nemoci.
 www.printo.it/pediatric-rheumatology/cz/intro Majeedův Verze č 2016 1. CO JE MAJEEDŮV SYNDROM? 1.1 Co je to? Majeedův syndrom je vzácné, geneticky podmíněné onemocnění. Pacienti mají projevy chronické
www.printo.it/pediatric-rheumatology/cz/intro Majeedův Verze č 2016 1. CO JE MAJEEDŮV SYNDROM? 1.1 Co je to? Majeedův syndrom je vzácné, geneticky podmíněné onemocnění. Pacienti mají projevy chronické
Charakteristiky vybraných deformit pátere Detská kyfóza Scheuermanova nemoc Hyperlordóza Plochá záda Skoliotické držení - skolióza
 DEFORMITY PÁTERE Obsah prednášky Charakteristiky vybraných deformit pátere Detská kyfóza Scheuermanova nemoc Hyperlordóza Plochá záda Skoliotické držení - skolióza Detská kyfóza (školní kulatá záda) Prícina:
DEFORMITY PÁTERE Obsah prednášky Charakteristiky vybraných deformit pátere Detská kyfóza Scheuermanova nemoc Hyperlordóza Plochá záda Skoliotické držení - skolióza Detská kyfóza (školní kulatá záda) Prícina:
HEMOFILIE - DIAGNOSTIKA A LÉČBA V SOUČASNOSTI
 HEMOFILIE - DIAGNOSTIKA A LÉČBA V SOUČASNOSTI Patofyziologie hemofilie Deficit koagulačního faktoru VIII (hemofilie A) nebo IX (hemofilie B), jeho dysfunkce nebo přítomnost jeho inhibitorů vede k poruše
HEMOFILIE - DIAGNOSTIKA A LÉČBA V SOUČASNOSTI Patofyziologie hemofilie Deficit koagulačního faktoru VIII (hemofilie A) nebo IX (hemofilie B), jeho dysfunkce nebo přítomnost jeho inhibitorů vede k poruše
Autor: Kouřilová H., Biolková V., Školitel: Šternberský J., MUDr. Klinika chorob kožních a pohlavních, LF UP v Olomouci
 Raynaudův fenomén Autor: Kouřilová H., Biolková V., Školitel: Šternberský J., MUDr. Klinika chorob kožních a pohlavních, LF UP v Olomouci Raynaudův fenomén je klinický stav, který je charakterizován občasnými
Raynaudův fenomén Autor: Kouřilová H., Biolková V., Školitel: Šternberský J., MUDr. Klinika chorob kožních a pohlavních, LF UP v Olomouci Raynaudův fenomén je klinický stav, který je charakterizován občasnými
Merosin deficitní kongenitální svalová dystrofie.
 Merosin deficitní kongenitální svalová dystrofie www.adamhrdy.cz Narození a určení diagnozy Adámek se narodil jako prvorozený syn, dne 19.8.2013 v Thomayerově nemocnici v Krči. Po narození nic nenasvědčovalo
Merosin deficitní kongenitální svalová dystrofie www.adamhrdy.cz Narození a určení diagnozy Adámek se narodil jako prvorozený syn, dne 19.8.2013 v Thomayerově nemocnici v Krči. Po narození nic nenasvědčovalo
Dědičnost vázaná na X chromosom
 12 Dědičnost vázaná na X chromosom EuroGentest - Volně přístupné webové stránky s informacemi o genetickém vyšetření (v angličtině). www.eurogentest.org Orphanet - Volně přístupné webové stránky s informacemi
12 Dědičnost vázaná na X chromosom EuroGentest - Volně přístupné webové stránky s informacemi o genetickém vyšetření (v angličtině). www.eurogentest.org Orphanet - Volně přístupné webové stránky s informacemi
Obr.1 Žilní splavy. https://s-media-cache-ak0.pinimg.com/564x/c3/91/8c/c3918c00db875bb460cf868b26ee1a0c.jpg
 TROMBÓZA NITROLEBNÍCH ŽIL A SPLAVŮ Autor: Barbora Baštinská Výskyt Mozková žilní trombóza je vzácné onemocnění, jehož příznaky se mohou značně lišit. Vyskytuje se spíše u mladších pacientů a většinou (až
TROMBÓZA NITROLEBNÍCH ŽIL A SPLAVŮ Autor: Barbora Baštinská Výskyt Mozková žilní trombóza je vzácné onemocnění, jehož příznaky se mohou značně lišit. Vyskytuje se spíše u mladších pacientů a většinou (až
V Ý R O Č N Í Z P R Á V A
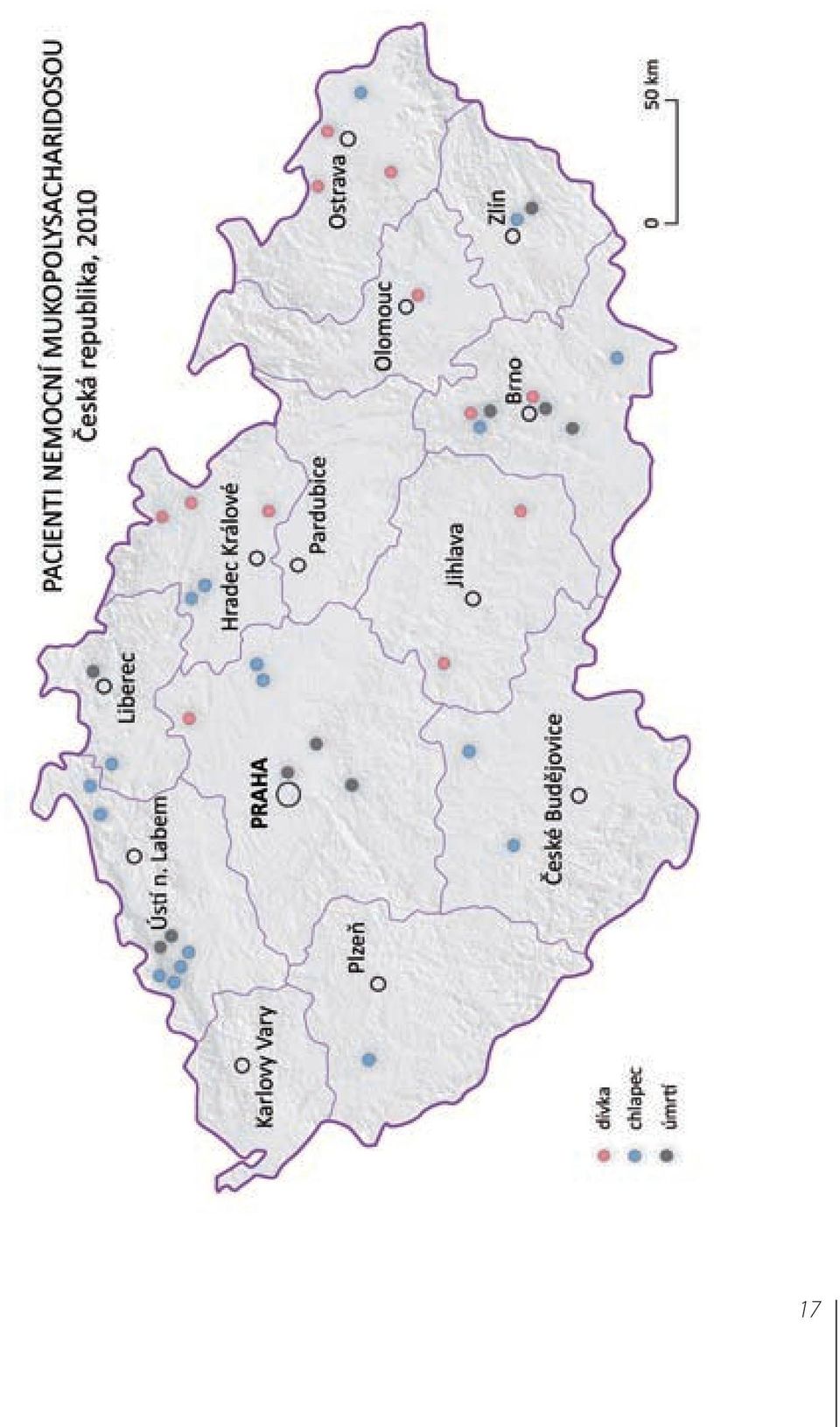
 Společnost pro mukopolysacharidosu (1994 2010) společenství rodin dětí nemocných nevyléčitelnou a smrtelnou nemocí V Ý R O Č N Í Z P R Á V A 2010 Chaloupky 35, 772 00 Olomouc, IČO: 61985112, bank.spojení:
Společnost pro mukopolysacharidosu (1994 2010) společenství rodin dětí nemocných nevyléčitelnou a smrtelnou nemocí V Ý R O Č N Í Z P R Á V A 2010 Chaloupky 35, 772 00 Olomouc, IČO: 61985112, bank.spojení:
Kapitola III. Poruchy mechanizmů imunity. buňka imunitního systému a infekce
 Kapitola III Poruchy mechanizmů imunity buňka imunitního systému a infekce Imunitní systém Zásadně nutný pro přežití Nezastupitelná úloha v obraně proti infekcím Poruchy imunitního systému při rozvoji
Kapitola III Poruchy mechanizmů imunity buňka imunitního systému a infekce Imunitní systém Zásadně nutný pro přežití Nezastupitelná úloha v obraně proti infekcím Poruchy imunitního systému při rozvoji
Klasifikace tělesných postižení podle doby vzniku
 VÝUKOVÝ MATERIÁL: VY_32_INOVACE_ DUM 1, S 20 JMÉNO AUTORA: DATUM VYTVOŘENÍ: 25.1. 2013 PRO ROČNÍK: OBORU: VZDĚLÁVACÍ OBLAST. TEMATICKÝ OKRUH: TÉMA: Bc. Blažena Nováková 2. ročník Předškolní a mimoškolní
VÝUKOVÝ MATERIÁL: VY_32_INOVACE_ DUM 1, S 20 JMÉNO AUTORA: DATUM VYTVOŘENÍ: 25.1. 2013 PRO ROČNÍK: OBORU: VZDĚLÁVACÍ OBLAST. TEMATICKÝ OKRUH: TÉMA: Bc. Blažena Nováková 2. ročník Předškolní a mimoškolní
Rozvoj vzdělávání žáků karvinských základních škol v oblasti cizích jazyků Registrační číslo projektu: CZ.1.07/1.1.07/02.0162
 ZŠ Určeno pro Sekce Předmět Rozvoj vzdělávání žáků karvinských základních škol v oblasti cizích jazyků Registrační číslo projektu: CZ.1.07/1.1.07/02.0162 Téma / kapitola Mendelova 2. stupeň Základní Zdravověda
ZŠ Určeno pro Sekce Předmět Rozvoj vzdělávání žáků karvinských základních škol v oblasti cizích jazyků Registrační číslo projektu: CZ.1.07/1.1.07/02.0162 Téma / kapitola Mendelova 2. stupeň Základní Zdravověda
Obsah a časové rozmezí všeobecné preventivní prohlídky dětí
 Obsah a časové rozmezí všeobecné preventivní prohlídky dětí Systém preventivních prohlídek v ČR je dlouhodobě propracovaný preventivní program, který umožňuje u Vašeho dítěte odhalit a včas léčit nejrůznější
Obsah a časové rozmezí všeobecné preventivní prohlídky dětí Systém preventivních prohlídek v ČR je dlouhodobě propracovaný preventivní program, který umožňuje u Vašeho dítěte odhalit a včas léčit nejrůznější
14. 1. 2013. Popis využití: Výukový materiál s úkoly pro žáky s využitím dataprojektoru,
 VY_32_INOVACE_PSYPS13260ZAP Výukový materiál v rámci projektu OPVK 1.5 Peníze středním školám Číslo projektu: CZ.1.07/1.5.00/34.0883 Název projektu: Rozvoj vzdělanosti Číslo šablony: III/2 Datum vytvoření:
VY_32_INOVACE_PSYPS13260ZAP Výukový materiál v rámci projektu OPVK 1.5 Peníze středním školám Číslo projektu: CZ.1.07/1.5.00/34.0883 Název projektu: Rozvoj vzdělanosti Číslo šablony: III/2 Datum vytvoření:
Nemoci opěrné soustavy
 Nemoci opěrné soustavy EU peníze středním školám Didaktický učební materiál Anotace Označení DUMU: VY_32_INOVACE_BI1.4 Předmět: Biologie Tematická oblast: Biologie člověka Autor: RNDr. Marta Najbertová
Nemoci opěrné soustavy EU peníze středním školám Didaktický učební materiál Anotace Označení DUMU: VY_32_INOVACE_BI1.4 Předmět: Biologie Tematická oblast: Biologie člověka Autor: RNDr. Marta Najbertová
59/1997 Sb. VYHLÁŠKA. Ministerstva zdravotnictví
 59/1997 Sb. VYHLÁŠKA Ministerstva zdravotnictví ze dne 13. března 1997, kterou se stanoví indikační seznam pro zdravotní péči v odborných dětských léčebnách Změna: 350/2008 Sb. Ministerstvo zdravotnictví
59/1997 Sb. VYHLÁŠKA Ministerstva zdravotnictví ze dne 13. března 1997, kterou se stanoví indikační seznam pro zdravotní péči v odborných dětských léčebnách Změna: 350/2008 Sb. Ministerstvo zdravotnictví
CUKROVKA /diabetes mellitus/
 CUKROVKA /diabetes mellitus/ CUKROVKA /diabetes mellitus/ Řadíme ji mezi neinfekční chronická onemocnění Na jejím vzniku se podílí nezdravý způsob života Významnou úlohu sehrává dědičnost Významným rizikovým
CUKROVKA /diabetes mellitus/ CUKROVKA /diabetes mellitus/ Řadíme ji mezi neinfekční chronická onemocnění Na jejím vzniku se podílí nezdravý způsob života Významnou úlohu sehrává dědičnost Významným rizikovým
Familiární středomořská (Mediterranean) horečka (Fever)
 horečka (Fever)") www.printo.it/pediatric-rheumatology/cz/intro Familiární středomořská (Mediterranean) horečka (Fever) Verze č 2016 2. DIAGNÓZA A LÉČBA 2.1 Jak se nemoc diagnostikuje? Obecně se uplatňuje následující postup:
www.printo.it/pediatric-rheumatology/cz/intro Familiární středomořská (Mediterranean) horečka (Fever) Verze č 2016 2. DIAGNÓZA A LÉČBA 2.1 Jak se nemoc diagnostikuje? Obecně se uplatňuje následující postup:
a) Sledovaný znak (nemoc) je podmíněn vždy jen jedním genem se dvěma alelami, mezi kterými je vztah úplné dominance.
 Sledovaný znak (nemoc) je podmíněn vždy jen jedním genem se dvěma alelami, mezi kterými je vztah úplné dominance.") GENEALOGIE (Genealogická metoda. Genealogické symboly. Rozbor rodokmenů. Základní typy dědičnosti.) ÚVOD Genealogie je základem genetického vyšetření člověka, jehož cílem je stanovení typu dědičnosti daného
GENEALOGIE (Genealogická metoda. Genealogické symboly. Rozbor rodokmenů. Základní typy dědičnosti.) ÚVOD Genealogie je základem genetického vyšetření člověka, jehož cílem je stanovení typu dědičnosti daného
Mutace genu pro Connexin 26 jako významná příčina nedoslýchavosti
 Mutace genu pro Connexin 26 jako významná příčina nedoslýchavosti Petr Lesný 1, Pavel Seeman 2, Daniel Groh 1 1 ORL klinika UK 2. LF a FN Motol Subkatedra dětské ORL IPVZ Přednosta doc. MUDr. Zdeněk Kabelka
Mutace genu pro Connexin 26 jako významná příčina nedoslýchavosti Petr Lesný 1, Pavel Seeman 2, Daniel Groh 1 1 ORL klinika UK 2. LF a FN Motol Subkatedra dětské ORL IPVZ Přednosta doc. MUDr. Zdeněk Kabelka
THANATOFORNÍ DYSPLAZIE I. TYPU. FN Ostrava Ivana Bubová Lucie Poloučková
 THANATOFORNÍ DYSPLAZIE I. TYPU FN Ostrava Ivana Bubová Lucie Poloučková THANATOFORNÍ DYSPLAZIE patří do skupiny kostních dysplazií neboli osteochondrodysplazií - dědičných chorob skeletu, které vznikají
THANATOFORNÍ DYSPLAZIE I. TYPU FN Ostrava Ivana Bubová Lucie Poloučková THANATOFORNÍ DYSPLAZIE patří do skupiny kostních dysplazií neboli osteochondrodysplazií - dědičných chorob skeletu, které vznikají
Chronická obstrukční plicní nemoc MUDR.ŠÁRKA BARTIZALOVÁ BARTIZALOVAS@FNPLZEN.CZ
 Chronická obstrukční plicní nemoc MUDR.ŠÁRKA BARTIZALOVÁ BARTIZALOVAS@FNPLZEN.CZ Nařízení vlády č. 114/2011 Platné od 1.7.2011 Kapitola III, položka 13 Chronická obstrukční plicní nemoc s FEV1/FVC méně
Chronická obstrukční plicní nemoc MUDR.ŠÁRKA BARTIZALOVÁ BARTIZALOVAS@FNPLZEN.CZ Nařízení vlády č. 114/2011 Platné od 1.7.2011 Kapitola III, položka 13 Chronická obstrukční plicní nemoc s FEV1/FVC méně
KLINICKÝ PŘÍNOS RADIOLOGICKÝCH PARAMETRŮ U SPONDYLOGENNÍ CERVIKÁLNÍ MYELOPATIE
 KLINICKÝ PŘÍNOS RADIOLOGICKÝCH PARAMETRŮ U SPONDYLOGENNÍ CERVIKÁLNÍ MYELOPATIE Neurologická klinika LF MU a FN Brno Tomáš Horák 7.12.2017 MYELOPATIE = nezánětlivé onemocnění míchy Spondylogenní (kompresivní)
KLINICKÝ PŘÍNOS RADIOLOGICKÝCH PARAMETRŮ U SPONDYLOGENNÍ CERVIKÁLNÍ MYELOPATIE Neurologická klinika LF MU a FN Brno Tomáš Horák 7.12.2017 MYELOPATIE = nezánětlivé onemocnění míchy Spondylogenní (kompresivní)
Nemoci nervové soustavy. Doc. MUDr. Otakar Keller, CSc.
 Nemoci nervové soustavy Doc. MUDr. Otakar Keller, CSc. MKN 10 - VI.kap.l G00-99 G00-G09 Zánětlivé nemoci centrální nervové soustavy G10-G13 Systémové atrofie postihující primárně nervovou soustavu G20-G26
Nemoci nervové soustavy Doc. MUDr. Otakar Keller, CSc. MKN 10 - VI.kap.l G00-99 G00-G09 Zánětlivé nemoci centrální nervové soustavy G10-G13 Systémové atrofie postihující primárně nervovou soustavu G20-G26
ROZDÍLOVÁ TABULKA NÁVRHU PRÁVNÍHO PŘEDPISU S PŘEDPISY EU
 V. ROZDÍLOVÁ TABULKA NÁVRHU PRÁVNÍHO PŘEDPISU S PŘEDPISY EU Rozdílová tabulka návrhu vyhlášky, kterou se mění vyhláška č. 277/2004 Sb., o stanovení zdravotní způsobilosti k řízení motorových vozidel, zdravotní
V. ROZDÍLOVÁ TABULKA NÁVRHU PRÁVNÍHO PŘEDPISU S PŘEDPISY EU Rozdílová tabulka návrhu vyhlášky, kterou se mění vyhláška č. 277/2004 Sb., o stanovení zdravotní způsobilosti k řízení motorových vozidel, zdravotní
Ošetřovatelské aspekty péče u pacientů s plicní arteriální hypertenzí léčených Remodulinem
 Ošetřovatelské aspekty péče u pacientů s plicní arteriální hypertenzí léčených Remodulinem Dagmar Hetclová 1. Interní klinika kardiologická, FN Olomouc Plicní arteriální hypertenze (PAH) plicní hypertenze
Ošetřovatelské aspekty péče u pacientů s plicní arteriální hypertenzí léčených Remodulinem Dagmar Hetclová 1. Interní klinika kardiologická, FN Olomouc Plicní arteriální hypertenze (PAH) plicní hypertenze
Hemofilie dnes. Investice do rozvoje a vzdělávání Operační program Vzdělávání pro konkurenceschopnost
 Hemofilie dnes Investice do rozvoje a vzdělávání Operační program Vzdělávání pro konkurenceschopnost Zdroje: Evropský sociální fond v ČR Evropská unie Ministerstvo školství, mládeže a tělovýchovy OPVK
Hemofilie dnes Investice do rozvoje a vzdělávání Operační program Vzdělávání pro konkurenceschopnost Zdroje: Evropský sociální fond v ČR Evropská unie Ministerstvo školství, mládeže a tělovýchovy OPVK
GUILLAIN BARÉ SYNDROM Z POHLEDU SESTRY. Autor: Kateřina Havelková Spoluautor: Silvia Pekárová
 GUILLAIN BARÉ SYNDROM Z POHLEDU SESTRY Autor: Kateřina Havelková Spoluautor: Silvia Pekárová Charakteristika onemocnění Jde o autoimunitní chorobu Je způsobené napadením myelinové pochvy nervů, axonů Provokujícím
GUILLAIN BARÉ SYNDROM Z POHLEDU SESTRY Autor: Kateřina Havelková Spoluautor: Silvia Pekárová Charakteristika onemocnění Jde o autoimunitní chorobu Je způsobené napadením myelinové pochvy nervů, axonů Provokujícím
DOWNŮV SYNDROM Úvod Downův syndrom trisomie Downův syndrom
 DOWNŮV SYNDROM Úvod Downův syndrom (DS), zvaný také jako Downova nemoc je nejrozšířenější formou mentální retardace. V literatuře se uvádí, že tímto syndromem je postiženo okolo 10% lidí s mentální retardací.
DOWNŮV SYNDROM Úvod Downův syndrom (DS), zvaný také jako Downova nemoc je nejrozšířenější formou mentální retardace. V literatuře se uvádí, že tímto syndromem je postiženo okolo 10% lidí s mentální retardací.
Kořenové syndromy. MUDr.Dana Vondráčková Centrum léčby bolesti FNB
 Kořenové syndromy MUDr.Dana Vondráčková Centrum léčby bolesti FNB Kořenové syndromy Cervikobrachiální syndrom Hrudní úžinový syndrom Výhřez bederní meziobratlové ploténky Pseudoradikulární bolesti Spondylolýza,
Kořenové syndromy MUDr.Dana Vondráčková Centrum léčby bolesti FNB Kořenové syndromy Cervikobrachiální syndrom Hrudní úžinový syndrom Výhřez bederní meziobratlové ploténky Pseudoradikulární bolesti Spondylolýza,
Huntingtonova choroba
 Huntingtonova choroba Renata Gaillyová OLG FN Brno Huntingtonova choroba je dědičné neurodegenerativní onemocnění mozku, které postihuje jedince obojího pohlaví příznaky se obvykle začínají objevovat mezi
Huntingtonova choroba Renata Gaillyová OLG FN Brno Huntingtonova choroba je dědičné neurodegenerativní onemocnění mozku, které postihuje jedince obojího pohlaví příznaky se obvykle začínají objevovat mezi
CADASIL. H. Vlášková, M. Boučková Hnízdová, A. Loužecká, M. Hřebíček, R. Matěj, M. Elleder
 CADASIL analýza mutací v genu NOTCH3 H. Vlášková, M. Boučková Hnízdová, A. Loužecká, M. Hřebíček, R. Matěj, M. Elleder Ústav dědičných metabolických poruch 1. LF UK a VFN Oddělení patologie a nár. ref.
CADASIL analýza mutací v genu NOTCH3 H. Vlášková, M. Boučková Hnízdová, A. Loužecká, M. Hřebíček, R. Matěj, M. Elleder Ústav dědičných metabolických poruch 1. LF UK a VFN Oddělení patologie a nár. ref.
TRÁPÍ VÁS NEBO VAŠE BLÍZKÉ BOLEST?
 TRÁPÍ VÁS NEBO VAŠE BLÍZKÉ BOLEST? ZDE APLIKUJEME -MD INJEKCE Kompletní řada certifikovaných zdravotnických středků terapii bolesti způsobené onemocněním pohybového a podpůrného aparátu. zmírnění bolesti
TRÁPÍ VÁS NEBO VAŠE BLÍZKÉ BOLEST? ZDE APLIKUJEME -MD INJEKCE Kompletní řada certifikovaných zdravotnických středků terapii bolesti způsobené onemocněním pohybového a podpůrného aparátu. zmírnění bolesti
http://www.vrozene-vady.cz
 Prevence vrozených vad z pohledu genetika MUDr. Vladimír Gregor, RNDr. Jiří Horáček odd. lékařské genetiky, Fakultní Thomayerova nemocnice v Praze Genetické poradenství Klinická genetika se zabývá diagnostikou
Prevence vrozených vad z pohledu genetika MUDr. Vladimír Gregor, RNDr. Jiří Horáček odd. lékařské genetiky, Fakultní Thomayerova nemocnice v Praze Genetické poradenství Klinická genetika se zabývá diagnostikou
ANAMNESTICKÝ ZDRAVOTNÍ DOTAZNÍK
 ANAMNESTICKÝ ZDRAVOTNÍ DOTAZNÍK Pro účely preventivní sportovně-kardiologické prohlídky ve zdravotnickém zařízení ProCorde s.r.o. v Chomutově. Příjmení, jméno:............................... Rodné číslo:.....................
ANAMNESTICKÝ ZDRAVOTNÍ DOTAZNÍK Pro účely preventivní sportovně-kardiologické prohlídky ve zdravotnickém zařízení ProCorde s.r.o. v Chomutově. Příjmení, jméno:............................... Rodné číslo:.....................
http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele
 http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
Varovné signály (Red flags) pro klinickou praxi vodítko pro zvýšené riziko genetické příčiny onemocnění u pacienta
 pro klinickou praxi vodítko pro zvýšené riziko genetické příčiny onemocnění u pacienta") Varovné signály (Red flags) pro klinickou praxi vodítko pro zvýšené riziko genetické příčiny onemocnění u pacienta Obecné varovné signály pro klinickou praxi Přítomnost jednoho nebo více varovných signálů
Varovné signály (Red flags) pro klinickou praxi vodítko pro zvýšené riziko genetické příčiny onemocnění u pacienta Obecné varovné signály pro klinickou praxi Přítomnost jednoho nebo více varovných signálů
Význam genetického vyšetření u pacientů s mentální retardací
 Význam genetického vyšetření u pacientů s mentální retardací Šantavá, A., Hyjánek, J., Čapková, P., Adamová, K., Vrtěl, R. Ústav lékařské genetiky a fetální medicíny FN a LF UP Olomouc Mentální retardace
Význam genetického vyšetření u pacientů s mentální retardací Šantavá, A., Hyjánek, J., Čapková, P., Adamová, K., Vrtěl, R. Ústav lékařské genetiky a fetální medicíny FN a LF UP Olomouc Mentální retardace
MUDr Zdeněk Pospíšil
 MUDr Zdeněk Pospíšil Imunita Charakteristika-soubor buněk,molekul a humorálních faktorů majících schopnost rozlišit cizorodé látky a odstranit je /rozeznává vlastní od cizích/ Zajišťuje-homeostazu,obranyschopnost
MUDr Zdeněk Pospíšil Imunita Charakteristika-soubor buněk,molekul a humorálních faktorů majících schopnost rozlišit cizorodé látky a odstranit je /rozeznává vlastní od cizích/ Zajišťuje-homeostazu,obranyschopnost
Výukový materiál zpracován v rámci projektu EU peníze školám
 http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
Rekurentní horečka spojená s NRLP21
 www.printo.it/pediatric-rheumatology/cz/intro Rekurentní horečka spojená s NRLP21 Verze č 2016 1. CO JE TO REKURENTNÍ HOREČKA SPOJENÁ S NRLP12 1.1 Co je to? Rekurentní horečka spojená s NRLP12 patří mezi
www.printo.it/pediatric-rheumatology/cz/intro Rekurentní horečka spojená s NRLP21 Verze č 2016 1. CO JE TO REKURENTNÍ HOREČKA SPOJENÁ S NRLP12 1.1 Co je to? Rekurentní horečka spojená s NRLP12 patří mezi
 www.printo.it/pediatric-rheumatology/cz/intro Blauův syndrom Verze č 2016 1. CO JE BLAUŮV SYNDROM/ JUVENILNÍ SARKOIDÓZA? 1.1 Co je to? Blauův syndrom je dědičné onemocnění. Pacienti trpí kombinací projevů:
www.printo.it/pediatric-rheumatology/cz/intro Blauův syndrom Verze č 2016 1. CO JE BLAUŮV SYNDROM/ JUVENILNÍ SARKOIDÓZA? 1.1 Co je to? Blauův syndrom je dědičné onemocnění. Pacienti trpí kombinací projevů:
Spinální svalová atrofie. Vypracovali: Kateřina Teplá Monika Madrová Anna Dobrovolná Mária Čižmárová Dominika Štrbová Juraj Štipka
 Spinální svalová atrofie Vypracovali: Kateřina Teplá Monika Madrová Anna Dobrovolná Mária Čižmárová Dominika Štrbová Juraj Štipka Klinický popis - relativně vzácná nemoc, nicméně se jedná o nejčastější
Spinální svalová atrofie Vypracovali: Kateřina Teplá Monika Madrová Anna Dobrovolná Mária Čižmárová Dominika Štrbová Juraj Štipka Klinický popis - relativně vzácná nemoc, nicméně se jedná o nejčastější
NEU/VC hodin praktických cvičení / blok
 Studijní program : Všeobecné lékařství Název předmětu : Neurologie Rozvrhová zkratka : NEU/VC012 Rozvrh výuky : 18 hodin seminářů / blok 72 hodin praktických cvičení / blok Zařazení výuky : 4. ročník,
Studijní program : Všeobecné lékařství Název předmětu : Neurologie Rozvrhová zkratka : NEU/VC012 Rozvrh výuky : 18 hodin seminářů / blok 72 hodin praktických cvičení / blok Zařazení výuky : 4. ročník,
Oftalmologie atestační otázky
 Platnost: od 1.1.2015 Oftalmologie atestační otázky Okruh všeobecná oftalmologie 1. Akomodace, presbyopie a její korekce 2. Refrakce oka, způsoby korekce, komplikace (mimo kontaktní čočky) 3. Kontaktní
Platnost: od 1.1.2015 Oftalmologie atestační otázky Okruh všeobecná oftalmologie 1. Akomodace, presbyopie a její korekce 2. Refrakce oka, způsoby korekce, komplikace (mimo kontaktní čočky) 3. Kontaktní
Rozhodovací proces při akutním útlaku míchy expansivním extraspinálním procesem
 Rozhodovací proces při akutním útlaku míchy expansivním extraspinálním procesem Zdeněk Adam a Pavel Šlampa Interní hematoonkologická klinika LF MU a FN Brno Radioterapeutická klinika MOU NADAČNÍ FOND Česká
Rozhodovací proces při akutním útlaku míchy expansivním extraspinálním procesem Zdeněk Adam a Pavel Šlampa Interní hematoonkologická klinika LF MU a FN Brno Radioterapeutická klinika MOU NADAČNÍ FOND Česká
Rozštěp neurální trubice. Klára Přichystalová Ondřej Sebera Jakub Ponížil Peter Salgó
 Rozštěp neurální trubice Klára Přichystalová Ondřej Sebera Jakub Ponížil Peter Salgó Rozdělení 1. Akranie/anencefalie akranie = chybění lebečního krytu s výhřezem mozkových struktur anencefalie = chybění
Rozštěp neurální trubice Klára Přichystalová Ondřej Sebera Jakub Ponížil Peter Salgó Rozdělení 1. Akranie/anencefalie akranie = chybění lebečního krytu s výhřezem mozkových struktur anencefalie = chybění
Maturitní témata. Předmět: Ošetřovatelství
 Maturitní témata Předmět: Ošetřovatelství 1. Ošetřovatelství jako vědní obor - charakteristika a základní rysy - stručný vývoj ošetřovatelství - významné historické osobnosti ošetřovatelství ve světě -
Maturitní témata Předmět: Ošetřovatelství 1. Ošetřovatelství jako vědní obor - charakteristika a základní rysy - stručný vývoj ošetřovatelství - významné historické osobnosti ošetřovatelství ve světě -
FAKULTA ZDRAVOTNICKÝCH STUDIÍ OŠETŘOVATELSKÁ PÉČE O NEMOCNÉ S MUKOPOLYSACHARIDÓZOU V DĚTSKÉM VĚKU
 FAKULTA ZDRAVOTNICKÝCH STUDIÍ Studijní program: Ošetřovatelství B5341 Anna Ţiţková Studijní obor: Všeobecná sestra 5341R009 OŠETŘOVATELSKÁ PÉČE O NEMOCNÉ S MUKOPOLYSACHARIDÓZOU V DĚTSKÉM VĚKU Bakalářská
FAKULTA ZDRAVOTNICKÝCH STUDIÍ Studijní program: Ošetřovatelství B5341 Anna Ţiţková Studijní obor: Všeobecná sestra 5341R009 OŠETŘOVATELSKÁ PÉČE O NEMOCNÉ S MUKOPOLYSACHARIDÓZOU V DĚTSKÉM VĚKU Bakalářská
- karyotyp: 47, XX, +18 nebo 47, XY, +18 = trizomie chromozomu 18 (po Downově syndromu druhou nejčatější trizomii)
") Edwardsův syndrom Edwardsův syndrom - karyotyp: 47, XX, +18 nebo 47, XY, +18 = trizomie chromozomu 18 (po Downově syndromu druhou nejčatější trizomii) - Prevalence v populaci: u narozených dětí cca 1:6500-1:8000,
Edwardsův syndrom Edwardsův syndrom - karyotyp: 47, XX, +18 nebo 47, XY, +18 = trizomie chromozomu 18 (po Downově syndromu druhou nejčatější trizomii) - Prevalence v populaci: u narozených dětí cca 1:6500-1:8000,
Míšní komprese u nádorů a úrazů páteře
 Míšní komprese u nádorů a úrazů páteře Chaloupka, R., Grosman, R., Repko, M., Tichý, V. Ortopedická klinika FN Brno-Bohunice Ortopedická klinika, FN Brno, Jihlavská 20, 625 00 Postižení páteře Bolest u
Míšní komprese u nádorů a úrazů páteře Chaloupka, R., Grosman, R., Repko, M., Tichý, V. Ortopedická klinika FN Brno-Bohunice Ortopedická klinika, FN Brno, Jihlavská 20, 625 00 Postižení páteře Bolest u
Tvorba elektronické studijní opory
 Záhlaví: Název studijního předmětu Téma Název kapitoly Autor - autoři Tvorba elektronické studijní opory Ošetřovatelská péče v neurologii Specifika ošetřovatelské péče u neurologických pacientů Specifika
Záhlaví: Název studijního předmětu Téma Název kapitoly Autor - autoři Tvorba elektronické studijní opory Ošetřovatelská péče v neurologii Specifika ošetřovatelské péče u neurologických pacientů Specifika
Diagnostika sluchových vad
 Klasifikace sluchových vad (opakování) a) místo vzniku postižení, b) doba vzniku postižení a c) stupeň postižení Základní pojmy z audiologie Sluchový práh Diagnostika sluchových vad - nejnižší intenzita
Klasifikace sluchových vad (opakování) a) místo vzniku postižení, b) doba vzniku postižení a c) stupeň postižení Základní pojmy z audiologie Sluchový práh Diagnostika sluchových vad - nejnižší intenzita
Jak se objednat na vyšetření?
 Jak se objednat na vyšetření? Ke genetické konzultaci nebo vyšetření v těhotenství odesílá praktický lékař, specialista nebo ošetřující gynekolog, který vystaví žádanku k vyšetření. Vyšetření provedená
Jak se objednat na vyšetření? Ke genetické konzultaci nebo vyšetření v těhotenství odesílá praktický lékař, specialista nebo ošetřující gynekolog, který vystaví žádanku k vyšetření. Vyšetření provedená
www.printo.it/pediatric-rheumatology/cz/intro
 www.printo.it/pediatric-rheumatology/cz/intro Candle Verze č 2016 1. CO JE CANDLE 1.1 Co je to? Chronická atypická neutrofilní dermatóza s lipodystrofií a zvýšenou teplotou (CANDLE) patří mezi vzácná dědičná
www.printo.it/pediatric-rheumatology/cz/intro Candle Verze č 2016 1. CO JE CANDLE 1.1 Co je to? Chronická atypická neutrofilní dermatóza s lipodystrofií a zvýšenou teplotou (CANDLE) patří mezi vzácná dědičná
Deficit mevalonátkinázy (MKD) (nebo hyper IgD syndrom)
 (nebo hyper IgD syndrom)") www.printo.it/pediatric-rheumatology/cz/intro Deficit mevalonátkinázy (MKD) (nebo hyper IgD syndrom) Verze č 2016 1. CO JE MKD? 1.1 Co je to? Deficit mevalonákinázy patří mezi dědičná onemocnění. Jedná
www.printo.it/pediatric-rheumatology/cz/intro Deficit mevalonátkinázy (MKD) (nebo hyper IgD syndrom) Verze č 2016 1. CO JE MKD? 1.1 Co je to? Deficit mevalonákinázy patří mezi dědičná onemocnění. Jedná
Šablona č. 01.33. Přírodopis. Opakování: Kosterní soustava člověka
 Šablona č. 01.33 Přírodopis Opakování: Kosterní soustava člověka Anotace: Opakování učiva o kosterní soustavě člověka Autor: Ing. Ivana Přikrylová Očekávaný výstup: Písemné opakování učiva o kosterní soustavě.
Šablona č. 01.33 Přírodopis Opakování: Kosterní soustava člověka Anotace: Opakování učiva o kosterní soustavě člověka Autor: Ing. Ivana Přikrylová Očekávaný výstup: Písemné opakování učiva o kosterní soustavě.
KOTVA CZ.1.07/1.4.00/21.3537
 KOTVA CZ.1.07/1.4.00/21.3537 Identifikátor materiálu EU: PRIR - 60 Anotace Autor Jazyk Vzdělávací oblast Vzdělávací obor PRIR = Oblast/Předmět Očekávaný výstup Speciální vzdělávací potřeby Prezentace žáka
KOTVA CZ.1.07/1.4.00/21.3537 Identifikátor materiálu EU: PRIR - 60 Anotace Autor Jazyk Vzdělávací oblast Vzdělávací obor PRIR = Oblast/Předmět Očekávaný výstup Speciální vzdělávací potřeby Prezentace žáka
Registry kloubních náhrad co všechno nám říkají
 Registry kloubních náhrad co všechno nám říkají Autor: Nieslaniková E., Školitel: Gallo J., prof. MUDr. Ph.D., Lošťák J., MUDr. Registry kloubních náhrad se zaměřují na získávání a shromažďování informací
Registry kloubních náhrad co všechno nám říkají Autor: Nieslaniková E., Školitel: Gallo J., prof. MUDr. Ph.D., Lošťák J., MUDr. Registry kloubních náhrad se zaměřují na získávání a shromažďování informací
Rozměr zavřeného průkazu mm
 Přední strana Str. 1 PRŮKAZ PACIENTA užívajícího přípravek Humira (určeno dospělým i dětským pacientům) Rozměr zavřeného průkazu 105 73 mm Je nutné předložit tuto kartičku každému lékaři či zdravotníkovi
Přední strana Str. 1 PRŮKAZ PACIENTA užívajícího přípravek Humira (určeno dospělým i dětským pacientům) Rozměr zavřeného průkazu 105 73 mm Je nutné předložit tuto kartičku každému lékaři či zdravotníkovi
Akutní jaterní porfýrie
 Akutní jaterní porfýrie CO DĚLAT MÁTE-LI PODEZŘENÍ NA ZÁCHVAT AKUTNÍ JATERNÍ PORFÝRIE? Mgr. Katarína Francová Orphan Drug Specialist Project Manager mobil: +420 702 211 762 e-mail: kfrancova@orphan-europe.com
Akutní jaterní porfýrie CO DĚLAT MÁTE-LI PODEZŘENÍ NA ZÁCHVAT AKUTNÍ JATERNÍ PORFÝRIE? Mgr. Katarína Francová Orphan Drug Specialist Project Manager mobil: +420 702 211 762 e-mail: kfrancova@orphan-europe.com
Státní zdravotní ústav Praha. Milovy 2017
 Alergie, KVO riziko Státní zdravotní ústav Praha Milovy 2017 Jana Kratěnová Spolupráce s 46 praktickými lékaři pro děti a dorost v 15 městech ČR Celkem 5130 dětí ve věku 5,9,13 a 17 let Data získána v
Alergie, KVO riziko Státní zdravotní ústav Praha Milovy 2017 Jana Kratěnová Spolupráce s 46 praktickými lékaři pro děti a dorost v 15 městech ČR Celkem 5130 dětí ve věku 5,9,13 a 17 let Data získána v
NÁROK NA PRŮKAZ OSOBY SE ZDRAVOTNÍM POSTIŽENÍM
 NÁROK NA PRŮKAZ OSOBY SE ZDRAVOTNÍM POSTIŽENÍM Aby mohl vzniknout nárok na průkaz, je nutné, aby z lékařských zpráv vyplývalo některé z následujících postižení. Je nutné, aby zpráva byla od lékaře, který
NÁROK NA PRŮKAZ OSOBY SE ZDRAVOTNÍM POSTIŽENÍM Aby mohl vzniknout nárok na průkaz, je nutné, aby z lékařských zpráv vyplývalo některé z následujících postižení. Je nutné, aby zpráva byla od lékaře, který
Sluch jako jeden ze základních pilířů mezilidské komunikace
 Sluch jako jeden ze základních pilířů mezilidské komunikace Jitka Vydrová Olga Bendová Lidská komunikace Vnímání a realizace řeči je náročný proces, který člověku zajišťuje místo v lidské společnosti.
Sluch jako jeden ze základních pilířů mezilidské komunikace Jitka Vydrová Olga Bendová Lidská komunikace Vnímání a realizace řeči je náročný proces, který člověku zajišťuje místo v lidské společnosti.
Akutní jaterní porfýrie
 Akutní jaterní porfýrie CO DĚLAT MÁTE-LI PODEZŘENÍ NA ZÁCHVAT AKUTNÍ JATERNÍ PORFÝRIE? Mgr. Katarína Francová Orphan Drug Specialist Project Manager mobil: +420 702 211 762 e-mail: kfrancova@orphan-europe.com
Akutní jaterní porfýrie CO DĚLAT MÁTE-LI PODEZŘENÍ NA ZÁCHVAT AKUTNÍ JATERNÍ PORFÝRIE? Mgr. Katarína Francová Orphan Drug Specialist Project Manager mobil: +420 702 211 762 e-mail: kfrancova@orphan-europe.com
von Willebrandova choroba Mgr. Jaroslava Machálková
 von Willebrandova choroba Mgr. Jaroslava Machálková von Willebrandova choroba -je dědičná krvácivá choroba způsobená vrozeným kvantitativním či kvalitativním defektem von Willebrandova faktoru postihuje
von Willebrandova choroba Mgr. Jaroslava Machálková von Willebrandova choroba -je dědičná krvácivá choroba způsobená vrozeným kvantitativním či kvalitativním defektem von Willebrandova faktoru postihuje
Downův syndrom. Renata Gaillyová OLG FN Brno
 Downův syndrom Renata Gaillyová OLG FN Brno Zastoupení genetických chorob a vývojových vad podle etiologie 0,6 %-0,7% populace má vrozenou chromosomovou aberaci incidence vážných monogenně podmíněných
Downův syndrom Renata Gaillyová OLG FN Brno Zastoupení genetických chorob a vývojových vad podle etiologie 0,6 %-0,7% populace má vrozenou chromosomovou aberaci incidence vážných monogenně podmíněných
Registrační číslo projektu: CZ.1.07/1.5.00/34.0649
 Výukový materiál zpracován v rámci projektu EU peníze školám Název školy: Střední zdravotnická škola a Obchodní akademie, Rumburk, příspěvková organizace Registrační číslo projektu: CZ.1.07/1.5.00/34.0649
Výukový materiál zpracován v rámci projektu EU peníze školám Název školy: Střední zdravotnická škola a Obchodní akademie, Rumburk, příspěvková organizace Registrační číslo projektu: CZ.1.07/1.5.00/34.0649
Operační a protetické možnosti léčení kostní nádorové bolesti
 Operační a protetické možnosti léčení kostní nádorové bolesti Chaloupka, R., Grosman, R., Repko, M., Tichý, V. Ortopedická klinika FN Brno-Bohunice Ortopedická klinika, FN Brno, Jihlavská 20, 625 00 Postižení
Operační a protetické možnosti léčení kostní nádorové bolesti Chaloupka, R., Grosman, R., Repko, M., Tichý, V. Ortopedická klinika FN Brno-Bohunice Ortopedická klinika, FN Brno, Jihlavská 20, 625 00 Postižení
Studie EHES - výsledky. MUDr. Kristýna Žejglicová
 Studie EHES - výsledky MUDr. Kristýna Žejglicová Výsledky studie EHES Zdroje dat Výsledky byly převáženy na demografickou strukturu populace ČR dle pohlaví, věku a vzdělání v roce šetření. Výsledky lékařského
Studie EHES - výsledky MUDr. Kristýna Žejglicová Výsledky studie EHES Zdroje dat Výsledky byly převáženy na demografickou strukturu populace ČR dle pohlaví, věku a vzdělání v roce šetření. Výsledky lékařského
Prof. MUDr. Jiří Vorlíček, CSc. Prof. MUDr. Jitka Abrahámová, DrSc. MUDr. Tomáš Büchler, PhD.
 Promítnutí pokroků lékařské vědy do funkčního hodnocení zdravotního stavu a pracovní schopnosti ve vztahu k mezinárodní klasifikaci nemocí a s přihlédnutím k Mezinárodní klasifikaci funkčních schopností
Promítnutí pokroků lékařské vědy do funkčního hodnocení zdravotního stavu a pracovní schopnosti ve vztahu k mezinárodní klasifikaci nemocí a s přihlédnutím k Mezinárodní klasifikaci funkčních schopností
Výukový materiál zpracován v rámci projektu EU peníze školám
 Výukový materiál zpracován v rámci projektu EU peníze školám Registrační číslo projektu: Šablona/číslo materiálu: Jméno autora: Třída/ročník CZ.1.07/1.5.00/34.0996 III/2 VY_32_INOVACE_TVD539 Mgr. Lucie
Výukový materiál zpracován v rámci projektu EU peníze školám Registrační číslo projektu: Šablona/číslo materiálu: Jméno autora: Třída/ročník CZ.1.07/1.5.00/34.0996 III/2 VY_32_INOVACE_TVD539 Mgr. Lucie
PRŮKAZ PACIENTA. užívajícího přípravek Imraldi (určeno dospělým i dětským pacientům) verze 1
 verze 1") PRŮKAZ PACIENTA užívajícího přípravek Imraldi (určeno dospělým i dětským pacientům) verze 1 Je nutné předložit tuto kartičku každému lékaři či zdravotníkovi při každé návštěvě zdravotnického zařízení.
PRŮKAZ PACIENTA užívajícího přípravek Imraldi (určeno dospělým i dětským pacientům) verze 1 Je nutné předložit tuto kartičku každému lékaři či zdravotníkovi při každé návštěvě zdravotnického zařízení.
Vrodené vývojové vady srdca. skupina 4
 Vrodené vývojové vady srdca skupina 4 -Vrozené srdeční vady (VSV) patří mezi nejčastější vrozené vývojové vady. Obecně tvoří vrozené vady oběhové soustavy více než 40 % všech registrovaných vrozených vad
Vrodené vývojové vady srdca skupina 4 -Vrozené srdeční vady (VSV) patří mezi nejčastější vrozené vývojové vady. Obecně tvoří vrozené vady oběhové soustavy více než 40 % všech registrovaných vrozených vad
Specifika traumatologie a chirurgie akutních stavů dětského věku Ladislav Plánka
 Specifika traumatologie a chirurgie akutních stavů dětského věku Ladislav Plánka Initial Assessment and Management Hlavní sdělení Identifikovat unikátní anatomické a fyzilogické charakteristiky dětí, které
Specifika traumatologie a chirurgie akutních stavů dětského věku Ladislav Plánka Initial Assessment and Management Hlavní sdělení Identifikovat unikátní anatomické a fyzilogické charakteristiky dětí, které
Zdravotní TV. Mgr. Jan Veverka a PaedDr. Jaroslav Dobýval
 Zdravotní TV Mgr. Jan Veverka a PaedDr. Jaroslav Dobýval Zdravotní tělesná výchova forma tělesné výchovy určená pro zdravotně oslabené jedince (z hlediska zdravotnické klasifikace se jedná o III. zdravotní
Zdravotní TV Mgr. Jan Veverka a PaedDr. Jaroslav Dobýval Zdravotní tělesná výchova forma tělesné výchovy určená pro zdravotně oslabené jedince (z hlediska zdravotnické klasifikace se jedná o III. zdravotní
Vybrané příklady spolupráce na návrhu klasifikačního systému CZ-DRG
 Vybrané příklady spolupráce na návrhu klasifikačního systému CZ-DRG Ladislav Plánka Česká pediatricko-chirurgická společnost ČLS JEP Fakultní nemocnice Brno Konference DRG Restart 2016 9. 11. 2016 Nevýhody
Vybrané příklady spolupráce na návrhu klasifikačního systému CZ-DRG Ladislav Plánka Česká pediatricko-chirurgická společnost ČLS JEP Fakultní nemocnice Brno Konference DRG Restart 2016 9. 11. 2016 Nevýhody
KBI/GENE Mgr. Zbyněk Houdek
 Genealogie KBI/GENE Mgr. Zbyněk Houdek Rodokmenové schéma Shromáždění informací o rodině je 1. důležitým krokem v genetickém poradenství. Rodokmenové schéma musí být srozumitelné a jednoznačné. Poskytuje
Genealogie KBI/GENE Mgr. Zbyněk Houdek Rodokmenové schéma Shromáždění informací o rodině je 1. důležitým krokem v genetickém poradenství. Rodokmenové schéma musí být srozumitelné a jednoznačné. Poskytuje
PAS v každodenní praxi dětské psychiatrie EVA ČÁPOVÁ
 PAS v každodenní praxi dětské psychiatrie EVA ČÁPOVÁ Poruchy autistického spektra Všepronikající hrubá neurovývojová porucha mozku PAS (autistic spektrum disorder ASD) 1979 Lorna Wing a Judith Gould Výskyt
PAS v každodenní praxi dětské psychiatrie EVA ČÁPOVÁ Poruchy autistického spektra Všepronikající hrubá neurovývojová porucha mozku PAS (autistic spektrum disorder ASD) 1979 Lorna Wing a Judith Gould Výskyt
Akutní axonální motorická neuropatie: kazuistika. Bálintová Z., Voháňka S. Neurologická klinika LF MU a FN Brno
 Akutní axonální motorická neuropatie: kazuistika. Bálintová Z., Voháňka S. Neurologická klinika LF MU a FN Brno Zánětlivé polyneuropatie multifokální či difúzní postižení periferního nervového systému
Akutní axonální motorická neuropatie: kazuistika. Bálintová Z., Voháňka S. Neurologická klinika LF MU a FN Brno Zánětlivé polyneuropatie multifokální či difúzní postižení periferního nervového systému
Nervová soustava je základním regulačním systémem organizmu psa. V organizmu plní základní funkce jako:
 Nervová soustava je základním regulačním systémem organizmu psa. V organizmu plní základní funkce jako: Přijímá podněty smyslovými orgány tzv. receptory (receptory), Kontroluje a poskytuje komplexní komunikační
Nervová soustava je základním regulačním systémem organizmu psa. V organizmu plní základní funkce jako: Přijímá podněty smyslovými orgány tzv. receptory (receptory), Kontroluje a poskytuje komplexní komunikační
Alergický pochod. Alergie v dětském věku- od atopického ekzému k respirační alergii
 Alergický pochod Alergie v dětském věku- od atopického ekzému k respirační alergii Kateřina Kopecká Centrum alergologie a klinické imunologie Nemocnice Na Homolce Things we knew, things we did Things we
Alergický pochod Alergie v dětském věku- od atopického ekzému k respirační alergii Kateřina Kopecká Centrum alergologie a klinické imunologie Nemocnice Na Homolce Things we knew, things we did Things we
3. Výdaje zdravotních pojišťoven
 3. Výdaje zdravotních pojišťoven Náklady sedmi zdravotních pojišťoven, které působí v současné době v České republice, tvořily v roce 2013 více než tři čtvrtiny všech výdajů na zdravotní péči. Z pohledu
3. Výdaje zdravotních pojišťoven Náklady sedmi zdravotních pojišťoven, které působí v současné době v České republice, tvořily v roce 2013 více než tři čtvrtiny všech výdajů na zdravotní péči. Z pohledu
SAMOSTATNÁ PRÁCE 2012 jmeno a prijmeni
 ZÁPADOČESKÁ UNIVERZITA V PLZNI SAMOSTATNÁ PRÁCE 2012 jmeno a prijmeni ZÁPADOČESKÁ UNIVERZITA V PLZNI Příušnice Samostatná práce Informatika a výpočetní technika KIV/IFYER jmeno a prijmeni Obsah 1 Příušnice
ZÁPADOČESKÁ UNIVERZITA V PLZNI SAMOSTATNÁ PRÁCE 2012 jmeno a prijmeni ZÁPADOČESKÁ UNIVERZITA V PLZNI Příušnice Samostatná práce Informatika a výpočetní technika KIV/IFYER jmeno a prijmeni Obsah 1 Příušnice