Obsah Předmluva Úvod do analýzy paliv... 5
|
|
|
- Květoslava Havlová
- před 6 lety
- Počet zobrazení:
Transkript
1 Obsah Předmluva Úvod do analýzy paliv... 5 Literatura Úvod do separačních metod Charakteristika a rozdělení separačních metod Termodynamika a účinnost separačního procesu Vybrané separační metody Destilace Extrakce Další vybrané separační metody Literatura Chromatografie v analýze paliv Teorie chromatografie Princip chromatografie Způsoby provedení chromatografické separace Rozdělení chromatografických technik Retenční parametry Mechanizmy chromatografické separace Teorie popisující vznik a tvar chromatografických zón Vyhodnocení a možnosti ovlivnění chromatografické separace Plynová chromatografie Princip plynové chromatografie Nosný plyn Injektory Kolony Detektory Aplikace plynové chromatografie v analýze paliv Kapalinová chromatografie Princip kapalinové chromatografie Literatura Spektroskopie v analýze paliv Úvod do spektroskopie Princip a základní rozdělení spektroskopických technik Vlastnosti elektromagnetického záření... 70
2 Kvalitativní a kvantitativní analýza Stanovení kovů a prvků Ultrafialová a viditelná spektroskopie (UV-vis) Úvod do UV-vis Luminiscenční spektroskopie Základní instrumentace Techniky měření Využití při analýze paliv a příbuzných produktů UV-vis spektroskopie souhrn Infračervená spektroskopie (IČ) Úvod do IČ Základní instrumentace Techniky měření Využití při analýze paliv a příbuzných produktů IČ spektroskopie souhrn Spektroskopie nukleární magnetické rezonance (NMR) Princip NMR Parametry NMR spektra Instrumentace NMR měření Základní charakteristiky interpretace NMR spekter Dvourozměrná NMR Využití NMR v analýze paliv a biopaliv Literatura Hmotnostní spektrometrie v analýze paliv Instrumentace Vakuový systém Vstup vzorku Iontový zdroj Iontová optika Analyzátor Detektor Ovládací a vyhodnocovací zařízení Interpretace hmotnostních spekter Hmotnostní spektrum Obecná pravidla pro interpretaci hmotnostních spekter
3 Hmotnostní spektra alkanů Hmotnostní spektra alkenů a cykloalkanů Hmotnostní spektra terpenů Hmotnostní spektra aromátů Hmotnostní spektra polyaromátů Hmotnostní spektra alkoholů Hmotnostní spektra esterů mastných kyselin Hmotnostní spektra halogenovaných uhlovodíků Pokročilé MS techniky Vícestupňová hmotnostní spektrometrie Spektrometrie pohyblivosti iontů Hmotnostní spektrometrie izotopických poměrů Hmotnostní spektrometrie s indukčně vázaným plazmatem Praktické aplikace hmotnostní spektrometrie v analýze paliv Literatura Termické metody v analýze paliv Elektrochemie v analýze paliv
4 PŘEDMLUVA Bude doplněno 4
5 1. ÚVOD DO ANALÝZY PALIV Autoři: Olga Pleyer, Violetta Pospelová Je zapotřebí značného úsilí ke správné charakterizaci chemických a fyzikálních vlastností uhlovodíkových sloučenin, surovin, frakcí a produktů s vysokým stupněm přesnosti. Nicméně komplexní analýza uhlovodíkových směsí je nezbytná k určení vlastností, které mohou pomoci při řešení problému v technologické praxi a optimalizaci výrobních technologií. Například, znalost složení surové ropy umožňuje rafinerii optimalizovat její konverzi na vysoce hodnotné produkty. Stejně tak je nezbytné určení vlastností, které popisují funkci a užitečnost hotového produktu, jeho neškodlivost vůči životnímu prostředí a zdraví člověka. Detailní znalost složení uhlovodíkových směsí umožňuje zvážit dopady na životní prostředí, přestože se do životního prostředí uvolňuje nepatrná část odpadů ze zpracování uhlovodíků. Kvůli velkému měřítku daných procesů je zde potenciál ovlivnit životní prostředí. Znalost složení je potom potřebná nejen k identifikaci zdrojů kontaminace, ale také k pochopení dopadů a účinků jeho potenciálně nebezpečných složek. Potřeba rozvoje analytických metod se během posledních tří desetiletí zvýšila kvůli změně složení vstupních surovin. Vzniklo to kvůli zvýšenému množství těžších surovin, které se nyní používají k výrobě kapalných produktů. Před energetickou krizí sedmdesátých let byly těžší suroviny zřídka používány jako zdroje kapalných paliv a používaly se k výrobě asfaltu. Nyní se tyto suroviny přeměňují v hodnotné kapalné produkty. Tyto vzorky jsou ze své podstaty o hodně složitější než vzorky z lehčích zdrojů ropy a představují nové výzvy pro analýzy v odvětvích výroby paliv a zpracování ropy. Navíc tyto těžké frakce obvykle obsahují vyšší podíl dusíku, síry a kyslíkatých sloučenin než lehčí frakce, což způsobuje problémy jak u rafinérských operací, tak i ve složení hotových výrobků. V důsledku toho byly vyvinuty různé analytické techniky pro separaci, identifikaci a kvantifikaci jednotlivých složek ve frakcích ropy. Analytické techniky, jimiž se zjišťuje charakter a složení uhlovodíkové směsi, lze rozdělit na normované metody a speciální instrumentální analýzy. Normované metody jsou předběžné testy poskytující obecné údaje o vzorcích a jsou založeny na jednoduchých zkouškách, jako je např. destilační křivka, obsah vody, hustota a obsah síry, které umožňují zaznamenat požadované nebo nežádoucí vlastnosti. Normované metody umožňují spolehlivé stanovení předepsaných fyzikálně-chemických kvalitativních parametrů vzorku většinou pomocí jednoúčelových přístrojů v krátkém čase a obvykle jsou založeny na rutinním postupu. Výsledek nemusí vždy odpovídat realitě, ale je vzájemně porovnatelný na různých pracovištích (např. destilační křivka ASTM D86). Tyto metody poskytují užitečný obecný obraz o kvalitě vzorku, nepokrývají však znalosti nezbytné k poskytnutí přiměřených údajů, například pro konstrukci zařízení nebo pro další využití vzorku. Proto se pro doplnění informace o chemickém složení a struktuře vzorku využívají tzv. speciální instrumentální analýzy. V tom případě se nemusí jednat o normovanou metodu, jedná se o individuálně vyvinuté analytické postupy kombinující několik kroků (předseparaci vzorku, zkoncentrování analytu, vlastní finální analytickou koncovku). Většinou je tato analýza časově náročná a jedná se o 5
6 kombinaci různých separačních a identifikačních technik. Tyto analytické postupy mohou být využity s cílem kvalitativní nebo kvantitativní analýzy. Kvalitativní analýza poskytuje dobrý přehled o složení vzorku. Její úkolem je zjistit, jaké složky (jednotlivé sloučeniny či skupiny látek) jsou ve vzorku přítomné. Provádí se postupem separace identifikace, např. pomocí kombinace chromatografie a hmotnostní spektrometrie. Někdy je postačující selektivní chemická reakce nebo spektrometrická metoda bez předchozí separace a zjednodušení vzorku. Kvalitativní analýza obvykle předchází kvantitativní analýze, protože kvantitativní analýza má za úkol nalézt kvantitativní vztahy mezi složkami zjištěnými při kvalitativním výzkumu. Pomocí kvantitativní analýzy lze určit množství analytu (jednotlivé sloučeniny či skupiny látek) ve vzorku. Provádí se obdobným postupem jako kvalitativní, avšak je nutná kalibrace odezvy detektoru pomocí vhodného standardu. Obtížnost kvantitativního stanovení také vzrůstá se stoupajícím bodem varu vzorku. Další problematické body analýzy komplexních uhlovodíkových směsí jsou: velký počet izomerních sloučenin, široké rozmezí molárních hmotností a bodů varu nutnost zjednodušení vzorku pomocí vhodné separační techniky (chromatografie), uhlovodíkové složky mají velmi podobný charakter fyzikálně-chemické vlastnosti se nemění skokově, ale spojitě ostré oddělení jednotlivých subfrakcí prakticky není možné, problematická identifikace jednotlivých typů uhlovodíků nežádoucí interference ve spektrech: n-alkany x iso-alkany, cykloalkany x alkeny, společný výskyt těkavých i netěkavých složek vzorku často nelze použít jeden univerzální analytický postup, ale je nutno volit několik různých postupů speciálně pro analýzu různých součástí vzorku. Z toho vyplývá, že u jednoduchých uhlovodíkových směsí je možná úplná kvantitativní analýza, tzv. detailní analýza, někdy i bez předchozí separace. Naopak, u jiných složitějších uhlovodíkových směsí není možné kvantitativní stanovení všech individuálních uhlovodíků, a proto se provádí např. stanovení uhlovodíkových skupin bez kvantitativního rozdělení skupiny na jednotlivé složky. Používá se několik základních typů analýz komplexních uhlovodíkových směsí: 1. Detailní analýza spočívá v kvalitativní a kvantitativní analýze individuálních složek v celém vzorku nebo v případě vybrané skupiny sloučenin (např. polyaromátů). Pro detailní analýzu zase se využívá postup separace identifikace. Pro separaci je nezbytná účinná separační technika (např. plynová nebo kapilární chromatografie) v kombinaci s vhodnou identifikační technikou (např. hmotnostní spektrometrie, UV-vis spektroskopie). Detailní analýzu komplexních vzorků s vysokým bodem varu lze provést obtížně (benzín, petrolej), v některých případech ani není možné (plynový olej a těžší frakce). 2. Skupinová analýza představuje stanovení obsahu sloučenin stejného charakteru a vlastností jako jedné skupiny (např. monoaromáty, diaromáty). Používá se v případě, že je vzorek natolik složitý, že nelze provést detailní analýzu nebo detailní analýza není nezbytně nutná. 6
7 Využívá se stejný princip separace identifikace, avšak nejsou již tak vysoké nároky na účinnou separační techniku, přímo lze využít i spektrální metody (hmotnostní spektrometrie, UV spektrometrie, FTIR). Lze ji využit pro větší rozsah bodů varu než u detailní analýzy, ale limitujícím faktorem je opět složitost (počet složek) vzorku. Je vhodná pro analýzu středních frakcí (motorová nafta, oleje). 3. Typová analýza je stanovení zastoupení typů uhlíku (aromatický, cyklanický, parafinický) v průměrné molekule vzorku. Provádí se bez separace vzorku, pouze pomocí vhodné spektrální metody (FTIR, nukleární magnetická rezonance). Používá se v případech, kdy nelze použít skupinovou analýzu, tzn. je vhodná pro analýzu nejtěžších uhlovodíkových frakcí (asfalt, dehet, zbytkové frakce). 4. Fingerprint (otisk palce) jednoduchý orientační profil vzorku chromatografický nebo spektrální, který lze porovnat s referenčním vzorkem. Poskytuje pouze kvalitativní informace pro rychlou screeningovou analýzu (lze usoudit na původ vzorku), podklad pro rozhodování o dalším způsobu analýzy. 5. Chemometrická analýza statistické zpracování velkého počtu naměřených analytických dat (hlavně FTIR a NIR) známých vzorků a jejich korelace s jejich změřenými fyzikálně-chemickými kvalitativními parametry. Umožňuje rychlé stanovení široké škály normovaných parametrů paliv (hustota, viskozita, bod vzplanutí, oktanové nebo cetanové číslo, destilační charakteristiky atd.). Přesnost analýzy závisí na velikosti kalibrační matrice vzorků a je důležité si uvědomit, že tyto korelační metody by se neměly používat k odhadu vlastností, které jsou mimo kalibraci. V současné době je snaha využit k řešení analytických problémů spojených s detailní analýzou komplexních uhlovodíkových směsí vícedimenzionální separace, tzn. sekvence separačních a analytických metod v několika stupních. LITERATURA R. B. Long, J. G. Speight: The composition of petroleum. Petroleum Chemistry and Refining, Taylor & Francis, J. Mostecký: Analýza uhlovodíkových surovin. Státní nakladatelství technické literatury, J. G. Speight: The Chemistry and Technology of Petroleum, Fifth Edition. Taylor & Francis, J. G. Speight: Handbook of petroleum product analysis. John Wiley & Sons,
8 2. ÚVOD DO SEPARAČNÍCH METOD Autoři: Martin Staš, Violetta Pospelová, Olga Pleyer Separační metody mají v chemii paliv velmi široké využití, ať už se jedná o industriální procesy zpracování paliv nebo jejich analýzu. Co se týče zpracování ropy, tak separační procesy jako destilace, extrakce, chromatografické dělení a jiné se uplatňují v průběhu celého rafinérského a případně petrochemického zpracování. Pro analýzu paliv a biopaliv jsou separační metody rovněž nezbytné. Cílem této kapitoly není poskytnout podrobný přehled všech různých separačních metod a ani diskutovat jejich využití při rafinérském zpracování ropy. Důraz je zde kladen na vybrané separační metody běžně používané k předúpravě vzorku před další analýzou, např. zakoncentrování nebo izolaci složky. Chromatografie a hmotnostní spektrometrie, které rovněž využívají separačních mechanismů, budou diskutovány v samostatných kapitolách Charakteristika a rozdělení separačních metod Separační metody se používají k dělení směsí na čisté složky nebo na skupiny složek se stejnými nebo podobnými fyzikálně-chemickými vlastnostmi. K základním charakteristikám separačních metod patří jejich selektivita, rozsah použitelnosti a frakcionační kapacita. Selektivita je schopnost separační metody dělit jednotlivé složky směsi na základě určité vlastnosti, např. na základě rozdílných bodů varů, molekulové hmotnosti, rozpustnosti, atd. Rozsah použitelnosti definuje schopnost separační metody dělit určitý typ vzorku, tedy pro jaký vzorek je daná metoda použitelná. Příkladem je schopnost dělit atomy, molekuly, ionty, případně schopnost dělit těkavé složky, atd. Frakcionační kapacita definuje maximální počet složek, které lze danou metodou dělit. Například plynová chromatografie na kapilárních kolonách umožňuje rozdělit stovky složek při jedné analýze. Separačních metod existuje velmi velký počet. Na základě principu dělení je lze rozdělit do dvou velkých skupin, a to na separační metody založené na (viz Tab. 2.1 a 2.2): fázových rovnováhách, rozdílech v rychlosti migrace. Tab. 2.1: Vybrané separační metody založené na fázových rovnováhách (převzato a upraveno z Churáček a kol. Analytická separace látek, viz seznam literatury) Rovnováha plyn-kapalina plyn-tuhá látka kapalina-kapalina kapalina-tuhá látka destilace sublimace extrakce srážení pěnové dělení plynová rozdělovací chromatografie separace na molekulových sítech plynová adsorpční chromatografie kapalinová rozdělovací chromatografie gelová permeační chromatografie frakční krystalizace separace na molekulových sítech zonální (zonové) tavení 8
9 Tab. 2.2: kapalinová adsorpční chromatografie iontově výměnná chromatografie Vybrané separační metody založené na rozdílech v rychlosti migrace (převzato a upraveno z Churáček a kol. Analytická separace látek, viz seznam literatury) polopropustná membrána ultrafiltrace obrácená (reverzní osmóza) dialýza elektrodialýza Migrační prostředí silové pole elektroforéza izotachoforéza ultracentrifugace frakcionace tokem termodifuze hmotnostní spektrometrie 2.2. Termodynamika a účinnost separačního procesu Separační metody jsou většinou založené na distribuci složek dělené směsi mezi dvě fáze. Pro libovolnou složku směsi (označme jako složka A) lze po dosažení rovnováhy tuto distribuci popsat distribuční konstantou KD. Distribuční konstanta je pro složku A definována jako podíl rovnovážných koncentrací složky A v obou fázích, viz rovnice 2.1. K D (A) = [A] 1 [A] 2 = n 1(A) V 2 V 1 n 2 (A) (2.1) V této rovnici symbol [A]i představuje rovnovážnou koncentraci složky A ve fázích 1 a 2, ni představuje látkové množství složky A ve fázích 1 a 2 a symbol Vi představuje objem fází 1 a 2. V případě chromatografie a extrakce existuje konvence, že v čitateli se uvádí rovnovážná koncentrace složky ve stacionární resp. organické fázi, ve jmenovateli pak rovnovážná koncentrace složky v mobilní resp. vodné fázi. Podíl látkových množství složky A v obou fázích se označuje jako kapacitní poměr k (nebo kapacitní faktor), viz rovnice 2.2. k(a) = n 1(A) n 2 (A) Spojením rovnic 2.1 a 2.2 lze distribuční konstantu uvést následovně, viz rovnice 2.3. (2.2) K D (A) = k(a) V 2 V 1 (2.3) Na to, aby složky mohly být příslušnou separační metodou založené na fázové rovnováze rozdělena, musí se lišit hodnotou své distribuční konstanty. Poměr distribučních konstant (resp. kapacitních faktorů) dvou složek se označuje jako separační faktor, viz rovnice
10 α = K D(A) K D (B) = k(a) k(b) (2.4) Dle konvence se dosazují hodnoty do rovnice 2.4 tak, aby měl separační faktor hodnotu vyšší než jedna. Separační faktor slouží k vyhodnocení separačního procesu. V praxi však má při hodnocení separačního procesu vetší význam veličina výtěžek složky R, který je definován rovnicí 2.5. R(A) = m 1 (A) m 1 (A) + m 2 (A) (2.5) V této rovnici představuje symbol mi hmotnost složky A v příslušné fázi a součet ve jmenovateli udává celkovou hmotnost složky A v systému. Spojením rovnic 2.5 a 2.2 lze výtěžek složky vyjádřit pomocí kapacitních poměrů, viz rovnice 2.6. R(A) = k(a) k(a) + 1 (2.6) 2.3. Vybrané separační metody Destilace Destilace je velmi často využívána fyzikální separační metoda, která umožňuje dělit složky kapalné směsi na základě různých bodů varu, resp. těkavosti. Bod varu je obecně definován jako teplota při daném tlaku, při které existuje termodynamická rovnováha mezi kapalnou a plynnou fází. Zahřátím k varu je část směsi převedena do plynné fáze, odděleně zkondenzuje, čím se získá destilát, který obsahuje těkavější složky. Zbylá část původní směsi se nazývá destilační zbytek. V destilačním zbytku se koncentrují méně těkavé a netěkavé složky. V případě analýzy směsí uhlovodíků se destilace používá ke: zúžení destilačního rozmezí na požadovanou destilační frakci, rozdělení uhlovodíkové směsi na několik užších frakcí, které jsou obvykle dále analyzovány kombinací dalších separačních a identifikačních metod, oddělení těkavých podílů od netěkavého zbytku, izolaci malých vzorků čistých uhlovodíků. Co se týče využití analytické destilace v analytice paliv, tak pro konvenční kapalná paliva se provádí tzv. destilační zkouška, která je popsána normou ČSN Jedná se o jednoduchou zkoušku prováděnou při atmosférickém tlaku na předepsané destilační aparatuře. Zaznamenávají se počáteční teplota (teplota, při které spadne první kapka kondenzátu do odměrného válce), teploty po předestilovaní určitého množství vzorku (např. 10, 20, 30 ml, atd.) a konečný bod varu (nejvyšší pozorovaná teplota). Výsledkem této zkoušky je destilační křivka, která vyjadřuje závislost předestilovaného množství testovaného paliva v objemových procentech na teplotě varu. Zkouška je reprodukovatelná a jednoduchá, nicméně začátek a konec destilace neodpovídá skutečným bodům varu. Kromě toho je k této zkoušce potřeba většího 10
11 množství vzorku (100 ml). Na základě destilační zkoušky je možné posoudit, o jaký typ paliva se jedná a zda není kontaminováno cizorodou frakcí. Navíc je možné z destilační zkoušky odhadnout i další parametry paliva (např. hustotu, bod vzplanutí apod.). Ke zjištění skutečných bodů je nevyhnutelné provádět destilaci v destilační aparatuře s účinností minimálně 100 teoretických pater. V současné době se k určení destilačního profilu paliv používá plynová chromatografie, přesněji technika simulované destilace (viz Obr. 2.1), která bude diskutována v kap Výhodou simulované destilace je zejména velice nízká spotřeba vzorku (řádově jednotky mikrolitrů a méně asfalt etrolej vakuový destilát (olejová frakce) Teplota varu ( C) ropa etrolej benzín etrolej plynový olej petrolej etrolej Předestilované množství (% hm.) Obr. 2.1: Destilační křivky simulované destilace ropy a různých ropných frakcí Teorie destilace a klasifikace destilačních procesů I když se v případě konvenčních paliv z ropy a biopaliv jedná o komplexní multikomponentní směsi, budeme pro další popis pro zjednodušení uvažovat binární, tedy dvousložkovou směs obsahující složky A a B, přičemž bude platit, že složka B má vyšší bod varu než složka A. Pro systém s ideálním chováním platí základní termodynamické zákony. Pro kapalnou fázi platí Raoultův zákon, viz rovnice 2.7 (uváděno pro složku A, stejný vztah lze vyjádřit také pro složku B). p(a) = x(a) p 0 (A) (2.7) V této rovnici je p(a) parciální tlak složky A nad kapalinou, x(a) je molární zlomek složky A v kapalině a p 0 (A) je tlak nasycených par čisté složky A při teplotě systému a normálním tlaku, tedy 101,325 kpa. 11
12 Obdobně pro plynnou fázi s chováním ideálního plynu platí Daltonův zákon vyjádřen rovnicí 2.8. p(a) = y(a) p (2.8) V této rovnici je p(a) parciální tlak složky A, y(a) je molární zlomek složky A v plynné fázi a p je celkový tlak systému, který se určí jako součet parciálních tlaků obou složek, viz rovnice 2.9. p = p(a) + p(b) = x(a) p 0 (A) + x(b) p 0 (B) (2.9) Spojením vztahů 2.7 a 2.8 se získá vztah popisující rovnováhu mezi plynnou a kapalnou fází, viz rovnice x(a) p 0 (A) = y(a) p y(a) = x(a) p0 (A) p (2.10) Destilaci lze vyhodnotit pomocí separačního faktoru, který se v destilaci nazývá relativní těkavost, viz rovnice 2.11, porovnej s rovnicí 2.4. α = K D(A) y(a) x(b) = K D (B) y(b) x(a) = p0 (A) p 0 (B) (2.11) Relativní těkavost je v ideální soustavě za stále teploty konstantní, závisí na poměru tlaků nasycených par čistých složek a nezávisí na poměru složek ve směsi. Je-li hodnota relativní těkavosti rovná jedné, směs nelze rozdělit. V reálných (neideálních) systémech se však projevují odchylky od ideálního chování, proto se do definice relativní těkavosti zavádí korekční faktory, kterými jsou aktivitní koeficienty složek A a B, viz rovnice Hodnota aktivitních koeficientů závisí na koncentraci, po korekci na neidealitu systému tak je i relativní těkavost funkcí poměru koncentrací obou složek. α = γ(a) p0 (A) γ(b) p 0 (B) (2.12) Podle způsobu provedení rozlišujeme tyto základní druhy destilace: jednoduchá destilace, rektifikace, destilace s inertní komponentou, extrakční destilace, azeotropická destilace, destilace za sníženého tlaku, molekulová destilace, mžiková destilace, termická rektifikace. 12
13 1. Jednoduchá destilace Využívá se při dělení látek s vysokým rozdílem v bodech varu nebo na hrubé rozdělení směsi. Jedná se o jednoduchou operaci nenáročnou na materiální vybavení. Pro směsi vroucí v úzkém rozmezí je však účinnost dělení často nedostatečná. 2. Rektifikace Rektifikace (nebo také protiproudá destilace) je destilační technika, která se vyznačuje výrazně vyšší separační účinnosti než jednoduchá destilace. Vyšší separační účinnost je dosažena díky zpětnému toku neboli refluxu. Ten je dosažen tak, že z kolony se odebírá pouze malá část destilátu a větší část se vrací zpět do kolony. Poměr látkového množství zpětného toku a látkového množství odebíraného destilátu za stejnou dobu se označuje jako poměr zpětného toku neboli refluxní poměr. V důsledku zpětného toku je počet ustanovení rovnováhy mezi kapalnou a plynnou fází mnohonásobně vyšší než u jednoduché destilace, čímž je daná také vyšší separační účinnost rektifikace. Účinnost rektifikace resp. dělící účinnost rektifikační kolony závisí na relativní těkavosti složek a počtu teoretických pater n destilační (srovnej s kap ). Parametr počet teoretických pater udává počet ustanovení rovnováhy mezi kapalnou a plynnou fází při průchodu kolonou. Čím je hodnota počet teoretických pater vyšší, tím je kolona účinnější. K porovnání kolon o různé délce byla zavedena veličina výškový ekvivalent teoretického patra H, viz rovnice Tato veličina udává délku kolony, která připadá na jedno ustanovení rovnováhy mezi plynnou a kapalnou fází. Z definice plyne, že čím je hodnota H nižší, tím je kolona účinnější. H = L n Rektifikaci lze provádět na několika typech kolon, které lze rozdělit následovně: (2.12) kolony s prázdnou trubicí: mají nižší separační účinnost (H = 5 15 cm) v porovnání s ostatními typy rektifikačních kolon, jelikož počet ustanovení rovnováhy mezi plynnou a kapalnou fází je zde nižší, kolony s výplní: ustavování rovnováhy se uskutečňuje na částicích výplně, H = 0,5 5 cm, kolony s rotující výplní: dosahují nejvyšších separačních účinností, H = 0,3 0,5 cm. 3. Destilace s přidanou komponentou Destilace s přidanou komponentou se používá za účelem zvýšení čistoty a výtěžku separovaných složek. Přidaná komponenta se obvykle volí tak, aby měla bod varu mezi body varů dělených složek. Lze však také použít vysokovroucí komponentu, která má vyšší bod varu než všechny dělené složky. Dále by přidaná komponenta měla být inertní a jednoduše oddělitelná od separovaných složek. Destilace s přidanou komponentou může pouze zlepšit destilační dělení směsi, která se v použité koloně dělí dobře. V případě, že použitá kolona na dělení nestačí, nelze rozdělení dosáhnout ani přídavkem inertní komponenty. Tento destilační postup se používá hlavně pro dělení organických sloučenin obsahujících heteroatom (dusík nebo kyslík), ale lze jej aplikovat také pro uhlovodíkové směsi. Příkladem 13
14 může být destilace směsi benzenu (bod varu 80 C) a toluenu (bod varu 111 C) s přídavkem propylacetátu (bod varu 101 C), kterou lze v porovnání s jednoduchou destilací dosáhnout vyšší čistotu benzenové a toluenové frakce a nižší obsah obou složek v mezifrakci. Kromě komponent vroucích mezi separovanými látkami lze využít i vysokovroucích komponent s vyšším bodem varu než nejvýše vroucí složka destilované směsi. Vybrané aplikace destilace s přidanou vysokovroucí komponentou jsou shrnuty níže. Destilace malého množství vzorku: přídavkem dostatečného množství vysokovroucí komponenty se zajistí vytěsnění i zbytků těžších podílů směsi, čímž se maximalizuje výtěžek destilátu. Destilace na koloně s malou zádrží: za účelem zvýšení výtěžku destilátu se volí přídavek vysokovroucí inertní komponenty s bodem varu minimálně o 50 C vyšší než nejtěžší složka, aby se minimalizovalo riziko kontaminace této složky přísadou. Destilace na koloně s velkou zádrží: v případě rektifikačních kolon s velkou zádrží se pro dosažení maximálního výtěžku destilátu používá vysokovroucí inertní komponenta s bodem varu o C vyšším, než je bod varu nejtěžší destilované složky. Přidaná komponenta tak vyplní celou rektifikační sekci, aniž by došlo k drastickým změnám v teplotním režimu kolony. Nebezpečí spočívá v tom, že přidaná látka může kontaminovat poslední dělenou frakci. 4. Extrakční destilace Princip extrakční destilace spočívá ve změně relativní těkavosti dělené směsi změnou poměru aktivitních koeficientů složek směsi v důsledku přídavku vhodného vysokovroucího rozpouštědla do rektifikační sekce. Vysokovroucí rozpouštědlo se při extrakční destilaci přidává kontinuálně do horní části destilační kolony a stéká dolů rektifikační sekcí proti proudu par destilujících uhlovodíků. Rozpouštědlo zůstává v kapalné fázi a hromadí se v destilační baňce. V okamžiku vydestilování lehčí složky do destilátu se vymění jímací válec a vydestiluje se výševroucí uhlovodíková složka a v destilační baňce zůstane jen rozpouštědlo. Extrakční destilace umožnuje dělení různých uhlovodíkových směsí o velmi mále relativní těkavosti (α: 1,0 1,2), ale i rozdělení směsí s poměrně blízkým bodem varu (10 20 C). Nejčastěji používána rozpouštědla v praxi jsou aceton, fenol, furfural, kresol, nitrobenzen, ale pro laboratorní účely je výběr vhodného rozpouštědla rozmanitější. Extrakční destilace má v rámci průmyslové separace veliký význam, ale pro analýzu uhlovodíkových směsí se využívá velmi zřídka. Vyžaduje složitější destilační aparaturu (kolonu s přívodem pro rozpouštědla) a mimo to je možné v rámci laboratorních podmínek dosáhnout podobných separačních výsledků i jinými jednoduššími laboratorními technikami, např. azeotropickou destilací, chromatografií, extrakcí, atd. 14
15 5. Azeotropická destilace Azeotropická destilace je založena na tvorbě azeotropu, což je směs dvou nebo více látek, které nelze destilací rozdělit, jelikož má tato směs stejné složení parní a kapalné fáze. Může se jednat buď o azeotrop s minimem bodu varu anebo azeotrop s maximem bodu varu, tedy azeotropická směs může být buď méně anebo více těkavá než čisté složky. Příčinou existence azeotropních směsí jsou interakce mezi molekulami směsi (např. vodíkové můstky). Azeotropická destilace umožnuje podobně jako extrakční destilace dělit směsi obsahující rozdílné strukturní typy, které vřou v úzkém rozmezí teplot, tedy mají blízké teploty varu. Laboratorní uspořádání pro azeotropickou destilaci se od běžné destilační aparatury nijak neliší. Princip azeotropické destilace spočívá v přídavku tzv. azeotropického činidla, které s jednou nebo několika složkami dělené směsi tvoří azeotrop s o minimálním bodu varu. Tím se zvětší rozdíly mezi body varu mezi dělenými složkami. Azeotropické činidlo se volí tak, aby jej bylo možné jednoduše odstranit, např. extrakcí, odstředěním. Vhodné je používat takové činidlo, které vře o 0 30 C méně než je bod varu dělené směsi. Mezi nejčastěji používaná azeotropická činidla patří různé alkoholy, fenoly, ketony, organické kyseliny, atd. Uhlovodíky mají sklon ke tvorbě azeotropických směsí, a to tím více, čím bližší jsou jejich body varu a čím více se od sebe liší v polaritě. Při analýze uhlovodíkových směsí je nezbytné s možnou tvorbou azeotropů počítat, a to zejména u úzkých frakcí, které obsahují nasycené a nenasycené uhlovodíky vedle sebe. 6. Destilace za sníženého tlaku Destilace za sníženého tlaku (nebo také vakuová destilace) využívá závislost bodu varu na tlaku. Se snižováním tlaku dochází rovněž ke snižování bodů varů sloučenin. Za sníženého tlaku tedy sloučeniny vřou při nižší teplotě, což lze s výhodou využít zejména pro vysokovroucí směsi, u kterých může v důsledku vysoké teploty docházet k rozkladu (krakování) části složek. Vedle toho má snížení tlaku vliv i na změnu relativní těkavosti, tím pádem je možné dosáhnout ostřejší separace složek než u destilace za atmosférického tlaku. 7. Destilace s vodní parou Destilace s vodní parou představuje specifický typ azeotropické destilace. Při tomto destilačním procesu se destilovanou směsi prohání vodní pára, která je vyvíjená v oddělené nádobě. Dělené složky se nesmí s vodou mísit. Vytvoření směsi destilovaných látek s vodní parou má za následek snížení bodu varu dělených složek. Tato destilace představuje šetrnou destilační metodu pro vysokovroucí sloučeniny. Pro uhlovodíkové směsi se tento typ destilace používá na šetrné oddělování netěkavých složek od uhlovodíkových podílů. 8. Mžiková destilace U klasické destilace jsou nejtěžší podíly po celou dobu destilace přítomné v destilační baňce a jsou tak vystaveny dlouhodobému působení zvýšené teploty, což může mít za následek tepelný rozklad destilovaných složek. Řešení tohoto problému je využití mžikové destilace. K destilaci se využívá aparatura s tzv. stékajícím rušeným filmem. Destilovaná kapalina je v aparatuře mechanicky na vyhřívané ploše rozrušována, čímž se docílí toho, že ze stékajícího filmu 15
16 okamžitě vypaří těkavé podíly, které zkondenzují v chladiči a odvedou se do primárního jímadla. Povrch filmu je neustále obnovován, což urychluje průběh destilace. Netěkavé podíly ze spodní části vyhřívané plochy jsou odváděny do druhého jímadla. Výhody této destilace jsou, že je vhodná pro vysokovroucí uhlovodíky s širokým destilačním rozmezím, probíhá jednak za atmosférického tlaku, v proudu inertního plynu, ale i za tlaku sníženého. 9. Molekulová destilace Molekulová destilace (nebo také destilace krátké dráhy) se provádí při tlaku kolem 0,1 Pa. V důsledku tohoto velmi nízkého tlaku je nad odpařovací plochou nízká koncentrace molekul vzduchu. Střední volná dráha destilovaných molekul se tedy významně prodlužuje, tedy snižuje se četnost jejich srážek s molekulami vzduchu a tím dochází ke zvýšení rychlosti vypařování a destilace. Aparatura pro molekulovou destilaci má kondenzační plochu v krátké vzdálenosti od výparné plochy a pracuje při tlaku, kdy střední volná dráha molekul přesáhne tuto vzdálenost. Molekulovou destilací mohou být předestilovány uhlovodíky až do C Extrakce Teorie extrakce a klasifikace extrakčních procesů Extrakce je separační proces, při kterém dochází k distribuci složky mezi dvě nemísitelné fáze. Tyto fáze mohou mít různé anebo stejné skupenství, viz tab Po dosažení rovnováhy lze distribuci popsat distribuční konstantou, viz rovnice 2.1, str. 9. U extrakce se místo pojmu distribuční konstanta používá pojem rozdělovací poměr. Rozdělovací poměr udává poměr koncentrace dělené složky v obou fázích. Složky směsi lze extrakcí rozdělit v případě, že separační faktor (viz rovnice 2.4, str. 10) se nerovná jedné, resp. je větší než jedna, jelikož separační faktor se dle konvence uvádí tak, aby nabýval hodnoty 1. Výtěžek extrakce lze vyjádřit pomocí vztahů 2.5 a 2.6, viz str. 10. Z rovnic 2.5 a 2.6 lze odvodit, že výtěžek extrakce a tedy množství analytu v kýžené fázi závisí na hodnotě rozdělovacího poměru a poměru obou extrakčních fází. Když extrakci opakujeme n-krát vždy s novou extrakční fázi, zvyšujeme tak výtěžek extrakce, což plyne z rovnice R n (A) = 1 (1 R(A)) n (2.13) Tab. 2.1: Přehled vybraných technik extrakce Typ extrakce Zkratka Popis a využití plyn-kapalina kapalina-kapalina GLE LLE Extrakce těkavých látek plynem z kapaliny. Poměrně často se používá se v plynové chromatografii pro zakoncentrování těkavých složek vzorku, viz headspace analýza, kap Založena na přechodu rozpuštěné látky z jedné kapalné fáze do druhé. Jedná se o velmi často používanou extrakční techniku, nicméně, dnes je již rychle vytlačována extrakcemi tuhou fází. tuhá látka-kapalina SLE Extrakce tuhých hlavně organických látek rozpouštědlem (loužení). 16
17 tuhá látka-tekutina v superkritickém stavu extrakce na tuhou fázi mikroextrakce na tuhou fázi SFE SPE SPME Extrakce tuhých hlavně organických látek tekutinou v superkritickém stavu. zachycení složek z kapalné nebo plynné fáze na tuhý sorbent, váže se buď analyt a nečistoty zůstanou v původní fázi anebo naopak Zachycení složek z kapalné nebo plynné fáze na tuhý sorbent, provádí se v mikroměřítku pomocí vlákna umístěného v mikrostříkačce, na kterém je nanesena vrstva sorbentu. K uvolnění (desorpci) navázaných složek dochází v důsledku zvýšené teploty, velice často se používá ve spojení s plynovou chromatografií, viz kap extrakce iontů kovů Látky v podobě iontů extrahovat nelze, vyžaduje se převod do elektroneutrální formy. Podle počtu ustavovaných rovnovah lze extrakční procesy rozdělit na procesy: jednostupňové: dochází pouze k jednomu ustanovení rovnováhy, příkladem je extrakce kapalina-kapalina v dělící baňce, mnohostupňové: ustavení rovnováhy se mnohonásobně opakuje v oddělených krocích, vyznačuje se vyšší separační účinností (výtěžek extrakce je vyšší) v porovnání s jednostupňovou extrakcí, zároveň se ale spotřebuje více extrakčního rozpouštědla, kontinuální: fáze jsou při protiproudném pohybu v neustálém styku. Dosahuje se tím vysokých separačních účinnosti při nižší spotřebě rozpouštědla než v případě mnohastupňové extrakce. Příkladem je extrakce v Soxhletově extraktoru. Vybrané extrakční procesy jsou blíže popsány níže Extrakce kapalina-kapalina Nejčastěji se provádí extrakce mezi dvě kapalné, vzájemně nemísitelné fáze. Tento typ extrakce se označuje extrakce kapalina-kapalina (LLE, z angl. liquid-liquid extraction) nebo také rozpouštědlová extrakce. Vzorek rozpuštěn v jedné kapalině je kvantitativně převeden do dělící baňky, do které je následně přidána druhá kapalina, která se s první kapalinou nemísí. Pro protřepání dělící baňky dochází k distribuci vzorku mezi dvě kapaliny a postupnému ustavení rovnováhy. Látky se rozdělují mezi tyto fáze na základě různé rozpustnosti v použitých rozpouštědlech. Čím větší je rozdíl v rozpustnosti, tím dokonalejší je oddělení. Cílem je selektivní oddělení analytu od ostatních složek nebo oddělení rušících látek od analytu. Část vzorku, která zůstává v původní kapalině, se nazývá rafinát. Část vzorku, která přechází do druhé kapaliny, se nazývá extrakt. Dle konvence se v čitateli rozdělovacího poměru (viz rovnice 2.1, str. 9) uvádí koncentrace složky ve fázi organické a ve jmenovateli koncentrace složky ve fázi vodní. Rozpouštědlová extrakce se obvykle používá, když samotná destilace nemůže poskytnout ekonomicky uspokojivé řešení, zejména v případě azeotropie nebo když je rozdíl v těkavosti složek směsi příliš malý. Pro uhlovodíkové směsi se tento typ extrakce používá zejména v níže uvedených případech. 17
18 Extrakce uhlovodíků z polárních médií (např. vodných vzorků). Používá se polární, málo polární, s vodou nemísitelné rozpouštědlo, např. n-pentan, n-hexan, CH2Cl2, cyklohexan, toluen, diethyléther, chloroform. N-hexan a cyklohexan jsou typické pro extrakci alifatických uhlovodíků. Dichlormethan a chloroform jsou typickými rozpouštědly pro nepolární až středně polární uhlovodíky (alifatické, aromatické, ethery, estery, aldehydy). Široká škála dostupných rozpouštědel pokrývá velké rozmezí rozpustnosti a selektivit, a je největší výhodou LLE. Extrakce uhlovodíků z nepolárních médií. Jako rozpouštědlo je vhodné použit polární, s uhlovodíky nemísitelné rozpouštědlo (dimethylsulfoxid, dimethylformamid, alkoholy). Lze provést extrakci relativně polárnějších aromátů a polyaromátů ze směsi alkanů a cykloalkanů, avšak částečně jsou extrahovány i nepolární alkany a cykloalkany Extrakce analytu z pevného vzorku K extrakci složek z pevného vzorku lze využít buď kapaliny, nebo tekutiny v superkritickém stavu. V prvním případě se jedná o extrakci tuhá látka-kapalina (SLE, z angl. solid-liquid extraction). SLE extrakce se může provádět v různých modifikacích, příkladem tohoto typu extrakce jsou Soxhletova extrakce, ultrazvuková extrakce a extrakce za vyššího tlaku. V druhém případě se jedná o extrakci tuhá látka-tekutina v superkritickém stavu (SFE, z angl. supercritical fluid extraction). Typickým příkladem SLE extrakce je Soxhletova extrakce. Jedná se o kontinuální extrakci za atmosférického tlaku, při které jsou extrakční fáze při protiproudém pohybu v neustálém kontaktu. Soxhletova extrakce se provádí v Soxhletově extraktoru, viz obr Vzorek je umístěn v papírové extrakční patroně s válcovým tvarem a kulatým dnem, která je umístěna ve střední části přístroje. Destilační baňka se naplní vhodným extrakčním rozpouštědlem, které dobře rozpouští dělenou složku nebo složky. Baňka je zahřívaná k varu rozpouštědla, jehož páry postupně stoupají postranní trubičkou, kondenzují na povrchu chladiče umístěného nad extrakční patronou se vzorkem a skapávají do patrony, kde se postupně hromadí a vymývají vzorek. Hladina rozpouštědla stoupá rovněž v tenké přepadové trubičce. Stoupne-li hladina rozpouštědla ve střední části extraktoru k nejvyšší části přepadové trubičky, přeteče roztok do destilační baňky. V destilační baňce se těkavé rozpouštědlo znovu zahřeje k varu a celý proces se kontinuálně opakuje. V destilační baňce se postupně hromadí dělená složka nebo složky. Získá se tak směs dělené složky nebo složek a extrakčního rozpouštědla, které se oddělí destilací. Výhodou tohoto procesu jsou vysoký extrakční výtěžek, malé nároky na přístrojové vybavení a díky kontinuálnímu provozu také nízká spotřeba rozpouštědla. Nevýhodou je délka separace, která se pohybuje obvykle řádově v hodinách. Tento typ extrakce rovněž není vhodný pro tepelně nestabilní sloučeniny. Dalším typickým příkladem SLE extrakce je ultrazvuková extrakce. Jedná se o extrakční proces, který je iniciován ultrazvukem. Aplikací ultrazvuku dochází k lepší penetraci rozpouštědla do vzorku a snadnějšímu uvolnění extrahovaných složek. Při ultrazvukové extrakci je tuhý vzorek ponořen do rozpouštědla (na dobu min) během které je aplikována ultrazvuková energie. Po skončení extrakce je extrakt od tuhého vzorku nebo jeho zbytků oddělen filtrací nebo 18
19 odstředěním a následně je odpařeno rozpouštědlo. Tento extrakční proces se obvykle provádí při laboratorní nebo mírně zvýšené teplotě, vzorek tedy není vůbec anebo jenom velmi málo tepelně namáhán. Zároveň se dosahují vysoké extrakční účinky a doba extrakce je krátká. Obvykle se však nejedná o kontinuální proces a vícestupňová ultrazvuková extrakce pak spotřebuje větší množství extrakčního rozpouštědla. Rozpustnost analytu v extrakčním rozpouštědle lze kromě použití ultrazvuku dosáhnout také zvýšením tlaku. Princip extrakce při vyšším tlaku spočívá v kombinaci zvýšené teploty ( C, obvykle víc než bod varu rozpouštědla) a tlaku (4 20 MPa) s použitím vhodného rozpouštědla (methanol, acetonitril, aceton, n-hexan apod.). Tyto podmínky zajištují zlepšení rozpustnosti analytu, zlepšují kontakt analytu s rozpouštědlem a zvyšují rychlost desorpce analytu z tuhé matrice do rozpouštědla. Výhodou je krátká doba extrakce a menší spotřeba rozpouštědla. Nevýhodou zase finanční náročnost aparatury. Analyt lze z pevného vzorku extrahovat rovněž tekutinou v superkritickém stavu, tedy při tlaku a teplotě nad kritickou hodnotou dané tekutiny. Tekutina v superkritickém stavu má fyzikálněchemické vlastnosti na rozmezí plynu a kapaliny. Kromě jiného se vyznačuje nízkou viskozitou a vysokou difuzivitou. Tyto vlastnosti umožňují rychlý přenos hmoty v důsledku příznivých charakteristik toku a snadné pronikání superkritické tekutiny do pórů pevné fáze a tím účinnou extrakci. Jako extrakční činidlo se nejčastěji využívá oxid uhličitý anebo směs oxidu uhličitého a methanolu. Výhodou SFE extrakce jsou krátká doba extrakce, možnost ovlivnit extrakci tlakem a snadná izolace analytu. Rovněž nedochází k nežádoucímu tepelnému namáhání vzorku a ztráty separovaných látek při odstraňování extrakčního činidla jsou minimální. Na druhé straně nevýhodou je vyšší finanční náročnost aparatury v porovnání s běžnými extrakčními procesy Extrakce a mikroextrakce na tuhou fázi Extrakce na tuhou fázi představuje extrakční metodu, při které se složky kapalného nebo také plynného vzorku váží na tuhý sorbent. Vázat se může buď analyt a matrice zůstává v původní fázi, nebo se vážou nečistoty (matrice) a v původní fázi setrvává analyt. Tato technika často slouží k předúpravě vzorku před další analýzou. Sorbent je obvykle umístěn v trubičce tvaru injekční stříkačky. SPE trubičky se většinou používají jednorázově, tedy neregenerují se. Jako sorbenty se používají běžné sorbenty používané v kapalinové chromatografii. Pro extrakci polárních sloučenin se používají silikagel, různě modifikovaný silikagel, alumina a florisil. Pro extrakci nepolárních sloučenin se používají uhlovodíky modifikovaný silikagel nebo aktivní uhlí. Pro extrakci iontů se používají iontoměniče. SPE extrakce je jednoduchá, relativně levná a díky široké škále sorbentů také široce využitelná. Tato technika ale není vhodná pro viskózní nebo velmi znečištěné vzorky, jelikož může docházet k ucpání SPE trubiček. Typický postup SPE extrakce SPE zahrnuje tyto kroky: kondicionace sorbentu: předúprava SPE kolonky vhodným rozpouštědlem, nanesení vzorku: dochází k zachycení analytu/ů, 19
20 promytí sorbentu: dochází k odstranění zbytků nečistot, aniž by byl eluován analyt, uvolnění analytu/ů: desorpce promytím z kolonky malým objemem vhodného rozpouštědla. SPE extrakci lze provádět taky v mikroměřítku. Mluvíme tehdy o mikroextrakci na tuhou fázi (SPME, z angl. solid phase micro-extraction). Jedná se o záchyt analytu z kapalné nebo plynné fáze na tenkém křemenném vlákně s chemicky vázanou fází. Účinnost SPME extrakce zaleží na několika faktorech, a to zejména na polaritě, tloušťce fáze nanesené na vlákno, způsobu vzorkování, teplotě vzorku a době extrakce, atd. SPME extrakce se používá často ve spojení s plynovou chromatografií. SPME vlákno je umístěno na držáku, který umožňuje vnést toto vlákno do nástřikového zařízení plynového chromatografu tak, aby vlákno nebylo poškozeno. Analyt se z SPME vlákna v nástřikovém zařízení desorbuje v důsledku zvýšené teploty Další vybrané separační metody Absorpce Princip absorpce je založený na přechodu cílených složek z plynné do kapalné fáze. Absorpce je využívána pro selektivní odstraňovaní nebo zakoncentrování stopového množství organických kontaminantů v plynných vzorcích, např. ve vzduchu. Když jsou požadované sloučeniny zředěny ve velkém množství plynu, zejména v nekondenzovatelných plynech, není vhodná klasická destilace, protože zajištění zpětného refluxu je velmi nákladné. Zde nastává vhodné použití absorpce za pomoci těžšího rozpouštědla. Výhodou absorpce je simultánní přechod cílových sloučenin ze vzorků různého původu do absorpčního roztoku. Navíc, absorpce v případě dobře vybraného rozpouštědla je vysoce selektivní. Nevýhodou je nemožnost odebrat reprezentativní vzorek pokud jsou v plynném vzorku přítomné částice aerosolu anebo tvrdé částice. Účinnost absorpce je závislá na rozpouštědle, koncentraci analytu, průtoku vzorkování, teplotě absorbéru, konstrukci absorbéru. Výběrem vhodného rozpouštědla lze zvýšit selektivitu na cílové analyty, např. absorpcí ve vodě lze oddělit C1-C4 alkoholy od jiných uhlovodíků, vyšší selektivity lze dosáhnout pomocí jiných polárních rozpouštědel jako dimethylformamid, n-butylacetát nebo polyethylenglykol. Např. nízké koncentrace C2-C4 uhlovodíků lze absorbovat do vody, uhličitanu sodného nebo methanolu. Pro izolaci stopových množství aromátů lze použit absorpci do kyseliny octové a následně neutralizaci alkalickým roztokem. Více informací o stanovení stopových množství uhlovodíků je uvedeno v kapitole Adsorpce Adsorpce je separační proces, jehož principem je hromadění plynné látky ze směsi plynů (adsorbátu) na povrchu pevné látky (adsorbentu) účinkem mezipovrchových přitažlivých sil. Rozlišují se dva druhy adsorpce, fyzikální adsorpce, která vzniká na základě Van der Waalsových přitažlivých sil a chemisorpce, která je tvořena pevnějšími chemickými vazbami. 20
21 Analytická adsorpce se používá pro záchyt organických sloučenin z plynných a vodných vzorků na vhodný sorpční materiál. Nejčastěji jsou využívány tzv. adsorpční trubičky. Princip metody spočívá v prostupu definovaného objemu plynné fáze přes trubičky naplněné vhodným sorbentem (aktivní uhlí, grafitizované molekulové síto (Carboxen), polymerní sorbenty (Tenax)). Poté následuje desorpce rozpouštědlem (CS2) nebo tepelná desorpce (do 350 C). Pokud se přes sorbent filtruje kapalný vzorek, tak se jedná o SPE extrakci, viz kap Frakční krystalizace Bude doplněno Separace na molekulových sítech Princip separace na molekulových sítech je založen na molekulově sítovém efektu. Na podobném principu je rovněž založena gelová permeační chromatografie. Molekulová síta jsou krystalické struktury s třírozměrným systémem pórů s přesně definovaným průměrem. Jako molekulová síta se používají syntetické zeolity (alumosilikáty). Molekuly s menší velikosti než je velikost pórů mohou pronikat do pórů a být adsorbovány, no molekuly větší než průměr pórů nikoliv. Molekulová síta se používají na odstraňování vody z plynů nebo kapalin a využívají se rovněž v plynové chromatografii na dělení permanentních plynů. Uplatnění nacházejí rovněž při dělení ropných frakcí zejména pro dělení nerozvětvených alkanů od ostatních typů uhlovodíků. Tato separace je však časově náročná, dělení probíhá řádově několik hodin, jelikož na to, aby dělené látky vnikly do pórů síta, se musí vhodně natočit. Další nevýhodou je obtížná separace zachycených nerozvětvených alkanů z molekulových sít. K uvolnění se používá buď extrakce vroucím n-heptanem po dobu 24 hodin, nebo rozložení sít kyselinami Sublimace Sublimaci lze považovat za zvláštní druh destilace. Jedná se o destilaci tuhé látky, při které teplota nesmí přesáhnout teplotu trojného bodu na p-t diagramu. Dochází tak k vypařování tuhé látky bez toho, aby se v systému objevila kapalina. Při zahřívání některých krystalických uhlovodíků se tlak nasycených par vyrovná s okolním tlakem dříve, než je dosažen bod tání. Takovéto uhlovodíky následně sublimují, tedy během zahřívání se vypařují do plynné fáze, aniž by látka roztála. Pro analýzu uhlovodíkových směsí se sublimace používá zejména k oddělení sublimujících uhlovodíku od nesublimujících nebo jako čistící operace. Pro sublimaci se využívají laboratorní sublimátory nebo aparatury na jednoduchou molekulovou destilaci. Měřítkem sublimovatelnosti látky je její sublimační bod, což je teplota, při níž se tlak nasycených par tuhé látky rovná vnějšímu tlaku. Teplota, při které se určitá sloučenina v laboratorních podmínkách sublimuje, je vždy nižší než její sublimační bod. Látky mohou sublimovat za atmosférického tlaku, za sníženého tlaku případně v proudu inertního plynu Zonální (zónové) tavení Zonální tavení je separační metoda, která se používá k dočištění tuhých látek získaných krystalizací nebo srážením. Základní podmínkou využitelnosti této metody je, aby byla čištěná 21
22 látka snadno tavitelná. Čištěná směs je vložena do vertikálně zavěšené trubice, která se pomalu spouští dolu prstencem ohřátým na teplotu, při které dochází k roztavení analytu. Po přechodu ohřívanou zónou analyt znovu ztuhne (vykrystalizuje), ale nečistoty zůstávají v kapalné formě v oblasti ohřívané zóny a postupně se hromadí v horní části trubice. Zonální tavení se používá k čištění některých krystalických uhlovodíků jako např. naftalen, difenyl, anthracen, pyren, atd. LITERATURA V. G. Berezkin, Y.S. Drugov: Gas chromatography in air pollution analysis, Elsevier, ISBN J. Churáček a kol.: Analytická separace látek. SNTL Nakladatelství technické literatury: Praha, Česká republika, ISBN: P. Klouda: Moderní analytické metody. Nakladatelství Pavel Klouda: Ostrava, Česká republika, ISBN: J. Mostecký, S. Hála, M. Kuraš, M. Popl: Analýza uhlovodíkových surovin. SNTL Nakladatelství technické literatury: Praha, Česká republika, M. Popl, J. Kubát: Separace látek. SNTL Nakladatelství technické literatury: Praha, Česká republika, L. Schreiberová a kol.: Chemické inženýrství I, 3. vydání. VŠCHT Praha: Praha, Česká republika, ISBN D. Sýkora: Separační metody výukové texty. VŠCHT Praha. J. P. Wauquier: Petroleum Refining, Volume 2 - Separation Processes. Editions Technip ISBN: K. Záruba a kol.: Analytická chemie, 1. díl. VŠCHT Praha: Praha, Česká republika, ISBN:
23 3. CHROMATOGRAFIE V ANALÝZE PALIV Paliva a biopaliva představují směsi, které je za účelem detailní nebo skupinové analýzy nutno vhodně rozdělit na čisté látky nebo na skupiny látek s podobnými vlastnostmi. Metodou první volby bývá často chromatografie, jež bude blíže popsána v této kapitole. Kapitola je rozdělená na tři základní části. V první části (kap. 3.1) bude popsána obecná teorie chromatografie. Druhá a třetí část (kap. 3.2 a 3.3) budou věnovány obecné charakteristice plynové, resp. kapalinové chromatografie. Obě tyto podkapitoly se rovněž budou věnovat praktickému využití těchto chromatografických technik v analýze paliv Teorie chromatografie Autor: Martin Staš Princip chromatografie Jak již bylo nastíněno v úvodu, chromatografie patří mezi separační techniky. V principu tedy umožňuje rozdělit směsi na jednotlivé složky (čisté látky) nebo na skupiny látek s podobnými fyzikálně-chemickými vlastnostmi. Podstatou chromatografické separace je interakce vzorku se dvěma fázemi, a to mobilní (pohyblivou) a stacionární (nepohyblivou). Mobilní fáze unáší vzorek (dělenou směs) stacionární fázi, která je obvykle uložená ve chromatografické koloně. Dochází přitom k opakovanému přenosu molekul vzorku mezi stacionární a mobilní fázi chromatografického systému. Tento systém se tak přibližuje rovnováze, kterou lze pro každou složku vzorku popsat distribuční konstantou KD, viz také kap Distribuční konstanta je pro libovolnou složku (označme složka A) vzorku definována jako podíl rovnovážných koncentrací této složky ve stacionární a mobilní fázi, viz rovnice 3.1. Složky vzorku mohou být v daném chromatografickém systému rozděleny pouze tehdy, když se liší hodnotou distribuční konstanty. Princip chromatografické separace totiž úzce souvisí se sílou interakce složek vzorku s chromatografickými fázemi. Čím ochotněji složka interaguje se stacionární fázi, tím déle je v koloně zadržována a tím je hodnota distribuční konstanty této složky vyšší. Naopak čím více složka interaguje s mobilní fázi, tím je zadržována méně a tím je její distribuční konstanta nižší. Aby bylo dosaženo dokonalého rozdělení vzorku na jednotlivé složky, musí být síla interakce těchto složek s fázemi chromatografického systému dostatečně odlišná. V takovémto případě se každá složka pohybuje kolonou svou vlastní rychlostí, která je dostatečně odlišná od rychlostí ostatních složek v daném vzorku. K D (A) = [A] s [A] m = n s(a) V m V s n m (A) (3.1) V této rovnici symbol [A]i představuje rovnovážnou koncentraci složky A ve stacionární a mobilní fázi, ni představuje látkové množství složky A v těchto fázích a symbol Vi představuje objem fází Způsoby provedení chromatografické separace Pro chromatografickou separaci existují tři základní způsoby provedení, a to: 23
24 eluční, vytěsňovací, frontální. Eluční chromatografie představuje chromatografické uspořádání, ve kterém je vzorek na kolonu nanášen v úzké zóně. Pro názornost si definujme, že vzorek je složen ze dvou složek složky A a složky B, viz Obr. 3.1a. Složky vzorku se váží na stacionární fázi a pro každou složku dochází k ustavení stavu blízkého rovnováze mezi složkou v mobilní a stacionární fázi. Prouděním mobilní fáze se tento stav poruší a složky přecházejí z fáze stacionární do fáze mobilní. Procesy ustavování a porušování rovnováhy se neustále opakují a složky se postupně opožďují vůči mobilní fázi. Zároveň vlivem toho, že síla interakce s chromatografickými fázemi je pro každou složku rozdílná, se tyto složky pohybují kolonou rozdílnou rychlostí a postupně se dělí. Kolonu opouští nejprve složka A a poté složka B. Složka B tedy stráví v koloně delší dobu než složka A. V ideálním případě dochází k dokonalému rozdělení vzorku a zóny obou složek jsou od sebe odděleny zónou čistého rozpouštědla. Platí, že šířka zón jednotlivých složek je větší než původní zóna vzorku. Rovněž platí, že šířka zóny složky B je větší než šířka zóny složky A (bude vysvětleno v kap ). Vytěsňovací chromatografie používá mobilní fáze, které jsou na stacionární fázi vázány silněji než jakákoliv složka vzorku. Mobilní fáze tak tlačí celý vzorek před sebou. Výsledný chromatogram tvoří stupně, přičemž zóny složek nejsou od sebe odděleny, tedy není mezi nimi zóna čisté mobilní fáze. Frontální chromatografie nepoužívá klasické mobilní fáze. Funkci mobilní fáze plní vzorek, který je k dispozici v dostatečném množství. Rozdělení vzorku není dokonalé, pouze první (nejméně zadržovaná) se získává čistá. Druhá složka se získává ve směsi s prvou, třetí ve směsi s první a druhou, atd. Výsledný chromatogram tvoří podobně jako v případě vytěsňovací chromatografie stupně. V tomto textu se budeme zabývat pouze eluční chromatografií, která představuje drtivou většinu všech chromatografických aplikací. Přístroj, kterým se provádí chromatografická separace, se označuje chromatograf. Časový záznam signálu detektoru, kterým prochází mobilní fáze z kolony, se nazývá chromatogram. Jednotlivé složky vzorku jsou ve chromatogramu zaznamenány ve formě chromatografických píků, viz Obr. 3.1b Rozdělení chromatografických technik Chromatografické techniky lze rozdělit podle skupenství mobilní a stacionární fáze, viz tab Mobilní fáze může být plynná nebo kapalná. Z toho plyne základní dělení chromatografie na plynovou a kapalinovou. Specifickým případem chromatografie z hlediska skupenství mobilní fáze je superkritická fluidní chromatografie, u které je mobilní fázi tekutina v superkritickém stavu. Stacionární fázi může tvořit kapalina vázána na tuhých částicích nebo na vnitřní stěně kolony, anebo se může jednat o tuhé porézní částečky o průměru jednotek až 24
25 stovek mikrometrů. Specifickým případem jsou stacionární fáze tvořeny jediným kusem pórovitého materiálu, který zcela vyplňuje vnitřní prostor kolony jedná se o tzv. monolitické stacionární fáze, které se využívají v kapalinové chromatografii. Tab. 3.1: Rozdělení chromatografických technik dle skupenství chromatografických fází Typ chromatografie Zkratka Mobilní fáze Stacionární fáze Plynová rozdělovací GLC plyn kapalina na nosiči Plynová adsorpční GSC plyn pevná látka Kapalinová rozdělovací LLC kapalina kapalina na nosiči Kapalinová adsorpční LSC kapalina pevná látka Iontově výměnná IEC kapalina pevná látka (ionex) Gelová permeační GPC kapalina pevná látka (gel) Superkritická fluidní SFC tekutina v superkritickém stavu kapalina na nosiči Techniky kapalinové chromatografie lze rovněž rozdělit dle způsobu uložení stacionární fáze na techniky sloupcové (kolonové) a plošné. V případě sloupcové chromatografie je stacionární fáze uložená ve chromatografické koloně. Chromatografické kolony jsou trubice ze skla, kovu nebo křemene, které jsou naplněné stacionární fázi. Rozměry kolony a způsob uložení stacionární fáze v koloně závisí od konkrétního typu chromatografie (bude specifikováno v kap. 3.2 a 3.3). V případě plošné chromatografie je stacionární fáze uložena na skleněné, hliníkové anebo plastové destičce. Nejčastějším příkladem tohoto typu chromatografických technik je tenkovrstvá chromatografie. Dále lze chromatografické techniky rozdělit na techniky analytické a preparativní. Analytické dělení se obecně provádí s velmi malými množstvími látek (obvykle jednotky miligramů a méně) za pomocí přístrojového vybavení. Preparativní separace se provádí s většími množstvími látek (desítky, stovky miligramů a více) a obvykle bez použití přístrojového vybavení Retenční parametry V kap byl v krátkosti vysvětlen princip chromatografické separace. Bylo zde uvedeno, že dochází k interakci složek vzorku s fází stacionární a mobilní. Sílou interakce libovolné složky A s chromatografickými fázemi je určena doba zdržení této složky v koloně, tj. její retenční čas tr. V chromatogramu je retenční čas složky určen průmětem maxima příslušného píku na časovou osu. Retenční čas se skládá z doby, kterou daná složka stráví v mobilní fázi (mrtvý retenční čas tm) a doby, kterou složka stráví ve fázi stacionární (redukovaný retenční čas t R), viz rovnice 3.2. Platí, že mrtvý retenční čas zadržované složky je stejný jako retenční čas složky, která v koloně zadržována není, tedy pohybuje se stejnou rychlostí jako mobilní fáze. Vynásobením retenčního času složky A průtokem mobilní fáze Fm se získá její retenční objem VR, viz rovnice 3.3. Obdobně platí, že vynásobením mrtvého retenčního času anebo redukovaného retenčního času se získají příslušné retenční objemy, a to mrtvý retenční objem VM 25
26 anebo redukovaný retenční objem V R, viz rovnice 3.4 a 3.5. Mrtvý retenční objem se přibližně rovná objemu mobilní fáze v koloně Vm. t R (A) = t M + t R(A) (3.2) V R (A) = F m t R (A) (3.3) V M = F m t M V m (3.4) V R(A) = F m t R(A) (3.5) Je-li L délka kolony, pak pro střední lineární rychlost mobilní fáze ū platí vztah 3.6 a pro lineární rychlost složky A u(a) pak platí vztah 3.7. u = L t M (3.6) u(a) = L t R (A) (3.7) Poměr lineární rychlosti složky A a střední lineární rychlost mobilní fáze se označuje jako retenční (retardační) faktor RF, viz rovnice 3.8. Z definice plyne, že hodnoty retenčního faktoru mohou nabývat hodnot v intervalu od nuly do jedné včetně mezních hodnot. Čím více se bude složka zdržovat ve stacionární fázi, tím více se bude hodnota retenčního faktoru blížit hodnotě 0. Naopak čím více se bude složka zdržovat ve fázi mobilní, tím více se bude retenční faktor blížit hodnotě 1. Mezních hodnot bude retenční faktor nabývat v případech kdy (i) složka neinteraguje se stacionární fázi a kolonou se pohybuje stejnou rychlostí jako mobilní fáze (hodnota 1), anebo (ii) složka neinteraguje s mobilní fází a je trvale zadržena na koloně (hodnota 0). Retenční faktor vyjadřuje pravděpodobnost, že se daná složka vyskytuje v mobilní resp. stacionární fázi. R F (A) = u(a) u = n M (A) n M (A) + n S (A) = t M t R (A) = k (3.8) k = n S(A) n M (A) = K D V S V m (3.9) Veličina k v rovnicích 3.8 a 3.9 se nazývá kapacitní faktor (nebo kapacitní poměr). Hodnotu kapacitního faktoru lze jednoduše vypočítat z rovnice k = t R(A) t M t M = V R(A) V M V M = V R(A) V M (3.10) Za předpokladu Vm = VM dostáváme rovnici 3.11, což je základní rovnice pro retenční objem. V R = V M + K D V S (3.11) V R = K D V S (3.12) 26
27 Mechanizmy chromatografické separace V chromatografické separaci se uplatňuje několik základních separačních mechanizmů, a to: adsorpce, rozdělování mezi dvě fáze (partiční proces), sítový efekt, iontová výměna (chemisorpce), bioafinita (specifické interakce biomolekul). Nicméně je nutno poznamenat, že v rámci kterékoliv chromatografické techniky dochází často k uplatnění několika z těchto mechanizmů současně. Většinou je však v rámci určité chromatografické techniky jeden z těchto mechanizmů dominantní. 1. Adsorpce je dominantním separačním mechanizmem v adsorpční chromatografii. Z chromatografických technik uvedených v tab. 3.1 se mezi techniky adsorpční chromatografie řadí GSC a LSC, tj. plynová a kapalinová chromatografie, kde stacionární fáze má tuhé skupenství. Může tedy docházet k adsorpci molekul složek vzorku z plynu nebo kapaliny na povrchu tuhého adsorbentu. Tyto molekuly jsou zachycovány v silovém poli na povrchu adsorbentu a na tomto povrchu setrvávají. Část molekul může překonat rozhraní tuhé fáze a pronikat do ní. Dochází tak k absorpci, což je konkurenční děj, který může probíhat současně s adsorpcí. U adsorpce rozlišujeme fyzikální adsorpci a chemisorpci. Podstatou fyzikální adsorpce je vytvoření van der Waalsových sil mezi tuhým adsorbentem a molekulami vzorku (adsorbátem). Energie těchto interakcí je velmi malá, ale rychlost jejich vytvoření je velká. Při chemisorpci se vytvářejí iontové nebo kovalentní vazby. Energie těchto interakcí je větší než v případě fyzikální adsorpce. Ke vzniku iontové nebo kovalentní vazby dochází pouze na aktivních centrech sorbentu. Kapacita povrchu sorbentu pro chemisorpci je tedy významně nižší než kapacita povrchu dostupná k fyzikální adsorpci. Rychlost vytvoření těchto interakcí je tak menší než v případě fyzikální adsorpce. K popisu adsorpční rovnováhy se používají adsorpční izotermy, které vyjadřují závislost rovnovážné koncentrace složky sorbované na tuhou fázi na rovnovážné koncentraci této složky ve fázi mobilní. Tyto isotermy jsou stejné při adsorpci z plynu nebo z roztoku. Tvar adsorpční isotermy ovlivňuje výsledný tvar chromatografického píku. V ideálním případě má adsorpční isoterma lineární průběh, čemuž odpovídá symetrický pík. V reálných systémech tento případ nastává spíše výjimečně, a to v oblasti velmi nízkých koncentrací. V reálných systémech má adsorpční isoterma tvar konvexní nebo konkávní. Tehdy mluvíme o Langmuirově resp. anti-langmuirově isotermě. V prvním případě dostáváme asymetrický pík protažený k vyšším retenčním časům pík takzvaně chvostuje. Ve druhém případě je symetrie píku opačná jedná se o tzv. frontující pík. V těchto dvou případech dostaneme symetrický pík pouze tehdy, když pracujeme v lineární částí Langmuirovy resp. anti-langmuirovy isotermy. Poslední typ adsorpční isotermy odpovídá chemisorpci. 2. Rozdělování mezi dvě fáze je dominantním separačním mechanizmem v GLC a LLC. U GLC se jedná o rozdělování (partiční proces) mezi plynnou mobilní a kapalnou stacionární fází. 27
28 Složky se dělí na základě jejich rozdílné rozpustnosti v kapalné stacionární fázi. Rozpustnost je ovlivněná těkavostí složek. Čím jsou složky těkavější, tím méně jsou na koloně zadržovány. Látky s podobnou těkavostí se dělí na základě svojí polarity. U LLC se jedná o partiční proces složky mezi dvě dokonale nemísitelné kapalné fáze. Hnací sílou tohoto procesu je koncentrační rozdíl složky v obou fázích chromatografického systému. 3. Sítový efekt je základním separačním mechanizmem u vylučovací chromatografie. Stacionární fází je gel, který má pórovitou strukturu. Molekuly vzorku jsou separovány na základě svojí velikosti. Platí, že čím je molekula menší, tím snáze vniká do pórů gelu a tím později opouští kolonu a naopak. 4. Iontová výměna je základním mechanizmem separace u iontově-výměnné chromatografie. Jedná se v podstatě o chemisorpci, kdy vhodný sorbent (iontoměnič) dokáže vázat určitý typ protiiontu a místo toho do roztoku uvolňovat jiný iont. 5. Bio-afinita je základním mechanizmem pro separaci bio-makromolekul, které lze jinak separovat jenom velmi obtížně, jelikož rozdíly v jejich fyzikálně-chemických vlastnostech jsou obvykle malé. Tento mechanismus se uplatňuje v kapalinové chromatografii. Princip je založený na tom, že se na vhodný nosič kovalentně naváže ligand, který je schopný vytvářet specifický komplex s určitou bio-makromolekulou, zatímco ostatní složky vzorku vázat nedokáže. Vytvořený komplex je reverzibilní a navázanou bio-makromolekulu lze z komplexu uvolnit použitím vhodného pufru nebo protiligandu Teorie popisující vznik a tvar chromatografických zón Bylo vypracováno několik teorií, které se zabývají studiem toho, proč v rámci chromatografické separace dochází k postupnému rozšiřování zón separovaných složek. V tomto textu budou blíže popsány rovnovážní teorie chromatografického patra a nerovnovážní (dynamická) van Deemterová teorie. Van Deemterová teorie vystihuje hlavně situaci, která nastává v plynové chromatografii. Bude proto přihlédnuto také k některým závěrům Giddingsové teorie, která lépe vystihuje situaci v kapalinové chromatografii Teorie chromatografického patra Teorii chromatografického patra zveřejnili poprvé Martin a Synge. Tato jednoduchá teorie je založená na pěti základních předpokladech. 1. Celá chromatografická kolona je hypoteticky rozdělena na velké množství elementárních jednotek chromatografických pater. 2. Na každém patře dochází k distribuci složky mezi mobilní a stacionární fázi a tato distribuce dosáhne rovnovážného stavu. 3. Hodnota distribuční konstanty je stejná ve všech patrech kolony. 4. Difuze ve směru toku je zanedbatelná. 5. Tok mobilní fáze je diskontinuální probíhá po malých přírůstcích, které mají objem jednoho patra. 28
29 Teorie chromatografického patra byla již záhy vystavena námitkám proti pěti základním předpokladům. Například zanedbání difuze ve směru toku mobilní fáze je neakceptovatelné zejména v plynové chromatografii. Rovněž diskontinualita průtoku je pro chromatografický proces nerealistická. V praxi se proto již teorie chromatografického patra nepoužívá. Nicméně tato teorie (rovněž jako teorie van Deemterová) vede k závěru, že tvar zóny vzorku a rovněž tvar příslušného chromatografického píku odpovídá Gaussově distribuci. Teorie chromatografického patra je srovnatelná s destilací. Dodnes se účinnost chromatografických i destilačních kolon udává pomocí počtu teoretických pater n pojmu, který plyne z této teorie. Účinnost chromatografické kolony lze kvantitativně vyjádřit pomocí veličin počet teoretických pater n a výškový ekvivalent teoretického patra (nebo výška teoretického patra) H. Mezi těmito veličinami platí vztah 3.13, přičemž L je délka kolony. Počet teoretických pater je definován vztahem H = L n (3.13) n = 16 t R 2 W b 2 = 5,54 2 t R 2 (3.14) W h/2 Čím je počet teoretických pater vyšší, tím vyšší je i účinnost kolony. Ze vztahu 3.14 plyne, že počet teoretických pater roste s klesající šířkou píku u základny a není pro danou kolonu konstantní, ale závisí na retenčním čase složky, která byla pro výpočet použita. Při uvádění hodnoty počtu teoretických pater by měla být rovněž uvedena délka kolony, aby bylo jasné, pro jakou kolonu bylo daného počtu pater dosaženo. Je tak možné určit výškový ekvivalent teoretického patra, který umožňuje porovnávat kolony o různé délce. Tato veličina má rozměr délky a představuje délku kolony, která připadá na jedno patro. V plynové chromatografii na kapilárních kolonách a v kapalinové chromatografii hodnoty počtu teoretických pater klesají s rostoucím retenčním časem složek. Proto se zavedla bezrozměrná veličina počet efektivních pater nef Teorie dynamická (van Deemterová) Van Deemterová teorie odráží skutečnost, že chromatografický systém se pouze blíží rovnováze, ale nikdy ji nedosáhne. Tento fakt je zapříčiněn skutečností, že v chromatografické koloně dochází ke kontinuálnímu proudění mobilní fáze, což zabraňuje dosažení rovnovážného stavu. Rozšiřování píků je dle van Deemterovy teorie zapříčiněno čtyřmi základními jevy, které budou blíže popsány níže. Tato teorie je vhodná, jak již bylo zmíněno, především pro popis situace v plynové chromatografii. 1. Vířivá difuze v mobilní fázi. Mobilní fáze proudí mezi částečkami pevného sorbentu (stacionární fáze), přičemž tyto částečky se mohou lišit svými rozměry a zároveň jsou uspořádány nepravidelně. Prostor mezi částečkami sorbentu, tedy kanálky, kterými proudí mobilní fáze, se liší svou velikostí. Rozšiřování 29
30 (rozmývání) zóny vzorku souvisí s tím, že ty molekuly vzorku, které proudí přímými kanálky, se kolonou pohybují rychleji než molekuly obtékající částečky sorbentu. H F = 2 λ d P (3.15) Příspěvek vířivé difuze k celkové výšce teoretického patra HF je přímo úměrná velikostí částic sorbentu dp a jejich tvaru. Tvar částice je charakterizován bezrozměrným geometrickým faktorem. Čím jsou částice sorbentu menší, tím menší je i příspěvek vířivé difuze k rozšiřování zóny vzorku. Platí totiž, že se zmenšujícím se průměrem částic stoupá homogenita náplně kolony a tím se zmenšují rozdíly ve velikostech kanálku. Tento příspěvek ne nezávislý na lineární rychlosti mobilní fáze. Pro kapilární kolony bez náplně je HF = Molekulární difuze v mobilní fázi. Zóna vzorku postupující kolonou je z obou stran obklopena zónou čisté mobilní fáze. Dle Fickova zákona dochází ke koncentračnímu gradientu, na základě čehož proudí molekuly vzorku (vyšší koncentrace) do zóny mobilní fáze (nižší koncentrace). K této difuzi dochází jak ve směru postupu, tak i proti směru postupu mobilní fáze. H L = 2 γ D m u (3.16) Příspěvek molekulární difuze HL je přímo úměrný velikosti difuzního koeficientu Dm složky a nepřímo úměrný lineární rychlosti mobilní fáze u v koloně. Symbol představuje bezrozměrný faktor. Jeho hodnota závisí na typu kolony. Pro náplňové kolony má hodnotu 0,6 a pro kapilární kolony je jeho hodnota 1. Hodnoty difuzních koeficientů jsou v kapalinové chromatografii přibližně o 4 až 5 řádů nižší než u plynové chromatografie. V kapalinové chromatografii se proto často příspěvek molekulární difuze zanedbává. 3. Odpor proti převodu hmoty ve stacionární fázi. Tento příspěvek se uplatňuje zejména v rozdělovací chromatografii (GLC a LLC), kde je kapalná stacionární fáze nanesena na nosiči nebo přímo na stěně kolony. Molekuly mohou pronikat do různé hloubky pod povrch stacionární fáze. Při zpětné desorpci jsou molekuly vzorku různě dlouho zadržovány, a to podle hloubky průniku do stacionární fáze. Molekuly pronikající pouze pod povrch stacionární fáze jsou zadržovány méně než hlouběji pronikající molekuly. H S = ψ d f 2 u D s k (k + 1) 2 (3.17) Příspěvek odporu proti převodu hmoty ve stacionární fázi HS je přímo úměrný lineární rychlosti mobilní fáze. Symbol je konfigurační faktor, který zohledňuje geometrii náplně, df je tloušťka filmu stacionární fáze, Ds je difuzní koeficient složky ve stacionární fázi, k je kapacitní faktor. V kapalinové adsorpční chromatografii a zejména pak v gelové chromatografii se lze setkat s pojmem stagnující mobilní fáze. Jedná se o mobilní fázi, která se nachází v pórech sorbentu 30
31 nebo gelu, tedy nepostupuje s hlavním tokem mobilní fáze, ale stagnuje. Molekuly, které pronikají do pórů gelu, a zdržují se tedy ve stagnující mobilní fázi, se opožďují za molekulami postupujícími v hlavním toku mobilní fáze. Příspěvek stagnující mobilní fáze na výšku teoretického patra HSM je přímo úměrný lineární rychlosti mobilní fáze. V grafu závislosti výšky teoretického patra na lineární rychlosti mobilní fáze budou tedy členy HS a HSM představovat přímky procházející počátkem. 4. Odpor proti převodu hmoty v mobilní fázi. H SM = d p 2 u k 30 D s (k + 1) (3.18) 2 Mobilní fáze proudící v kanálcích mezi částicemi sorbentu má v důsledku viskozity nižší rychlost v blízkosti povrchu sorbentu než v prostředku kanálků. V těsné blízkosti povrchu sorbentu je rychlost nulová. Molekuly vzorku unášeného kolonou proudem mobilní fáze se vůči sobě opožďují v závislosti od toho, kterým proudem jsou unášeny. Tento jev je částečně kompenzován difuzí molekul vzorku kolmo na směr proudění. H M = ω d p 2 u D m (3.19) Příspěvek odporu proti převodu hmoty ve fázi mobilní je přímo úměrný lineární rychlosti mobilní fáze. Symbol představuje bezrozměrný faktor, který charakterizuje typ náplně kolony. Význam zbylých symbolů již byl v této kapitole vysvětlen. Van Deemterův dynamický model předpokládá aditivní příspěvek výše zmíněných jevů k výškovému ekvivalentu teoretického patra. Tyto příspěvky se v jednotlivých chromatografických technikách uplatňují různou mírou (bude objasněno v kap. 3.2 a 3.3). H = H F + H L + H S + H M + H SM (3.20) V praxi se rovnice 3.20 uvádí spíše bez členu HSM, který má hlavní význam pouze v gelové chromatografii. Van Deemterova rovnice se rovněž obvykle vyjadřuje v tvaru vyjádřeném rovnicí 3.21, který vyjadřuje závislost H na střední lineární rychlosti mobilní fáze u. H = A + B u + C u + D u (3.21) Van Deemterová teorie dobře vystihuje situace, které nastávají v plynové chromatografii. Kapalinovou chromatografii lépe charakterizuje Giddingsova rovnice, viz rovnice Z Giddingsovy rovnice plyne, že vliv vířivé difuze a odporu proti přenosu hmoty jsou vzájemně závislé. V Giddingsově rovnici se zároveň zanedbává vliv molekulární difuze (HL), jelikož hodnota difúzních koeficientů v kapalinové chromatografii je o několik řádů menší než v chromatografii plynové. 1 H = 1 H H S F H M (3.22) 31
32 Vyhodnocení a možnosti ovlivnění chromatografické separace Cílem chromatografické separace je dosažení rozdělení vzorku na jednotlivé složky. Toto rozdělení může být úplné anebo neúplné. Pro kvantitativní popis chromatografického dělení lze použít dvě veličiny, a to separační faktor a (chromatografické) rozlišení. Separační faktor je pro dvě složky (A a B) definován jako podíl jejich rovnovážných konstant, resp. kapacitních faktorů těchto složek resp. jako podíl jejich redukovaných retenčních časů, viz rovnice = K D(B) K D (A) = k(b) k(a) = t R(B) t R(A) (3.23) Chromatografické rozlišení je pro dvě složky definováno pomocí jejich retenčních časů a šířek jejich píků u základny. R = t R (B) t R (A) 0,5 (W b (A) + W b (B) (3.24) Tento vztah lze přepsat do následovné podoby: R = N 4 α 1 α k(b) k(b) + 1 (3.25) Až rovnice 3.25 říká, které parametry je nutno ovlivnit, aby bylo dosaženo zvýšení chromatografického rozlišení a tím účinnější separace složek. Tento vztah obsahuje tři členy: kinetický, termodynamický a kapacitní. Z kinetického člena N/4 plyne, že rozlišení je přímo druhé odmocnině účinnosti kolony. Ovlivnění kinetického člena skutečné rovnice rozlišení má za následek změnu šířky píků. Mluvíme také o ovlivnění kinetického aspektu separace. Zlepšení chromatografického rozlišení ovlivněním kinetického aspektu separace tedy nastává v důsledku zúžení píků analytů. Všechny parametry ovlivňující kinetický aspekt separace jsou shrnuty v rovnicích van Deemterově a Giddingsově, viz kap Jedná se o tyto parametry: rychlost průtoku mobilní fáze, délka kolony, velikost částic sorbentu, difuzní koeficienty, atd. Z termodynamického člena rovnice (α 1)/α plyne, že rozlišení závisí také na separačním faktoru. Když se hodnoty separačního faktoru blíží jedné, rozlišení se blíží nule. Mluvíme také o ovlivnění termodynamického aspektu separace. Zlepšení chromatografického rozlišení ovlivněním termodynamického aspektu separace nastává v důsledku změny síly interakce složek vzorku s chromatografickými fázemi, což se projeví změnou jejich retenčních časů. Jednotlivé parametry ovlivňující termodynamický aspekt separace lze odvodit ze základní rovnice pro retenční objem, viz rovnice 3.11, str. 24. V této rovnici vystupují tři parametry, a to mrtvý retenční objem VM, distribuční konstanta KD a objem stacionární fáze VS. Mrtvý retenční 32
33 objem je z hlediska ovlivňování termodynamiky chromatografické separace nevýznamný, protože tento parametr je stejný pro všechny dělené složky. Naopak distribuční konstanta má na termodynamiku separace klíčový vliv. Distribuční konstantu lze ovlivnit změnou teploty separace a také změnou separačního mechanismu. Zvýšení teploty separace má za následek zvýšení kinetické energie molekul separovaných složek a tím redukci sil, které tyto složky zadržují v určité chromatografické fázi. Vliv teploty se uplatňuje zejména v plynové chromatografii. Separační mechanismy, které se mohou uplatňovat v chromatografii, byly diskutovány v kap Změnu separačního mechanismu a tím distribuční konstanty a termodynamického aspektu separace lze docílit následovně: změnou stacionární fáze (platí pro plynovou i kapalinovou chromatografii), změnou mobilní fáze (platí hlavně pro kapalinovou chromatografii). Posledním parametrem základní rovnice pro retenční objem je objem stacionární fáze. Zvýšení objemu (množství) stacionární fáze podporuje retenci jednotlivých dělených složek a tím také úspěšnost jejich vzájemného oddělení. Zvýšením objemu stacionární fáze v koloně rovněž dochází ke zvětšení kapacity kolony, s čím souvisí možnost nastřikovat větší množství vzorku. Na druhé straně je zvýšení objemu stacionární fáze spojeno s prodloužením doby analýzy a zhoršením kinetiky separace. Z kapacitního člena k(b)/(k(b) + 1) plyne, že rozlišení se zvyšuje s rostoucí hodnotou kapacitního faktoru. Když se kapacitní faktor blíží nule, kapacitní faktor se rovněž blíží nule. S kapacitním členem úzce souvisí frakcionační kapacita chromatografického systému, které udává maximální počet složek, které lze v daném systému separovat. Hodnotu kapacitního členu lze ovlivnit: množstvím stacionární fáze v koloně, změnou teploty (hlavně pro plynovou chromatografii), změnou stacionární a/nebo mobilní fáze. 33
34 3.2. Plynová chromatografie Autoři: Pavel Šimáček, Martin Staš Princip plynové chromatografie Plynová chromatografie (GC, z angl. gas chromatography) je chromatografická (separační) technika, kde mobilní fází je inertní plyn. Separovat lze permanentní plyny, kapaliny a rovněž pevné vzorky rozpuštěné ve vhodném rozpouštědle. Jelikož je mobilní fázi plyn, prvním krokem po zavedení vzorku do plynového chromatografu je jeho převedení do plynné fáze, a to zahřátím na teplotu v rozmezí přibližně C v závislosti na povaze vzorku. Z toho také plynou omezení plynové chromatografie. Podmínkou úspěšné plynově chromatografické analýzy je alespoň částečná těkavost vzorku. Vzorky zcela netěkavé (např. polymerní sloučeniny) plynovou chromatografií analyzovat nelze. Plynová chromatografie se vyznačuje velmi vysokou separační účinností, a to zejména ve spojení s kapilárními analytickými kolonami, které umožňují provádět detailní analýzu i pro vzorky obsahující stovky složek (např. automobilový benzin). U více komplexních vzorků již sice detailní analýza není proveditelná, i přesto je však plynová chromatografie zdrojem velmi cenných strukturních informací i pro takovéto vzorky. Plynové chromatografy pozůstávají z následovných základních součástí: zdroj nosného plynu, nástřikový člen (injektor), chromatografická kolona, detektor, počítač Nosný plyn Mobilní fází v plynové chromatografii je plyn, který unáší molekuly vzorku kolonou. Proto se pro něj vžil název nosný plyn. Nosný plyn je skladován v tlakových láhvích, ze kterých je do plynového chromatografu distribuován obvykle kovovými kapilárami. Jako nosný plyn se v plynové chromatografii nejčastěji používá dusík, helium, argon nebo vodík. Volba a také požadovaná čistota nosného plynu je ovlivněna zejména účelem analýzy a také typem použitého detektoru. Vlastnosti plynu, na které se přihlíží, jsou kromě čistoty inertnost, bezpečnost, hustota a viskozita. V plynové chromatografii jsou požadovány plyny o vysoké čistotě, obvykle více než 99,9995 %. Některé detektory (např. detektor elektronového záchytu) mají požadavky na čistotu nosného plynu ještě vyšší. Případné nečistoty by totiž mohly mít negativní vliv na analýzu, např. kyslík při vyšších teplotách může způsobovat oxidaci některých složek vzorku nebo stacionární fáze, uhlovodíky mohou ovlivňovat signál některých detektorů, voda může ovlivnit stacionární fázi nebo detektor, atd. Před vstupem do plynového chromatografu se nosný plyn proto ještě obvykle suší molekulovými síty a rovněž se zbavuje dalších nežádoucích nečistot průchodem přes 34
35 vhodný sorbent, ale i jinými způsoby např. kyslík z dusíku se odstraňuje přechodem kolonkou naplněnou mědí, kyslík z vodíku lze odstranit oxidací na platinovém katalyzátoru, uhlovodíky se odstraňují sorpcí na aktivním uhlí, atd. Ze závislosti výškového ekvivalentu teoretického patra na lineární rychlosti (průtoku) plynu plyne, že při nižších průtocích nosného plynu se maximální separační účinnosti kolony dosáhne použitím nosného plynu s větší hustotou (dusík, argon). Naopak když je potřeba pracovat při vyšších průtocích nosného plynu, je výhodné použít nosný plyn s menší hustotou (vodík, helium) Regulace tlaku a průtoku nosného plynu Bude doplněno Injektory Injektor neboli nástřikový člen je část plynového chromatografu, kterou do něj vzorek vstupuje. Mezi nejběžnější nástřikové členy patří: nástřikový člen s děličem toku, nástřikový člen PTV, nástřikový člen pro přímý nástřik na kolonu ( on column ). Při plynově-chromatografické analýze paliv se používá nejčastěji nástřikový člen s děličem toku. Tento nástřikový člen může pracovat ve dvou režimech, a to buď s otevřeným (split) anebo s uzavřeným (splitless) děličem. Používá se pro něj anglická zkratka SSL (neboli split/splitless). Vzorek je dávkovaný mikrostříkačkou do vyhřívaného výparníku, jehož teplota se obvykle pohybuje mezi C, a to v závislosti na těkavosti vzorku. Za těchto podmínek dochází k odpaření vzorku. Následně je proudem nosného plynu na kolonu vedena buď část vzorku (režim s otevřeným děličem) anebo celý vzorek (režim s uzavřeným děličem). Při režimu s otevřeným děličem je část vzorku vedena tokem nosného plynu otevřeným děličem mimo nástřikový člen a posléze mimo plynový chromatograf. Poměr množství vzorku, které se dostane na kolonu vs. množství vzorku, které se na kolonu nedostane, je určen poměrem množství nosného plynu vedeného na kolonu a mimo nástřikový člen, tedy tzv. splitovacím poměrem. Režim s otevřeným děličem se používá zejména v kombinaci s kapilárními analytickými kolonami, jež se vyznačují nízkými kapacitami, tedy jsou náchylné na přetížení (předávkování) a znečištění. Na tyto kolony tedy lze dávkovat pouze velmi malá množství vzorku. Režim s otevřeným děličem tak chrání kolonu před předávkováním a také možným znečištěním. Platí, že pro více znečištěné vzorky se volí vyšší splitovací poměry, případně je ještě vhodné tyto vzorky před analýzou naředit ve vhodném rozpouštědle. Režim s uzavřeným děličem se používá obvykle pouze při speciálním stanovení látek, které jsou ve vzorku přítomné ve stopovém množství (tzv. stopová analýza). Objem kapalných vzorků dávkovaných do injektoru typu SSL, za nímž je instalována kapilární kolona, bývá obvykle v rozmezí 0,1 1 µl. Hlavní výhodou tohoto nástřikového členu je možnost dvou režimů (univerzálnost) a cenová dostupnost. Hlavní nevýhodou je zejména možná diskriminace těžších (netěkavých) podílů, které mohou 35
36 kondenzovat v nástřikovém členu nebo na povrchu jehly dávkovací mikrostříkačky a nedostanou se tak na kolonu. Modernější obdoba klasického nástřikového členu je pak nástřikový člen typu PTV (z angl. programmed temperature vaporizer), u kterého lze navíc programovat teplotu vypařování vzorku, což rozšiřuje možnosti jeho využití. Do jisté míry tak může nástřik typu PTV zastat funkci klasického nástřiku typu SSL i níže popsaného nástřiku typu on-column. Při některých analýzách je někdy výhodné použít nástřikový člen typu on-column (OC) nazývaný též cool on-column (COC). U tohoto nástřiku je vzorek (zpravidla rozpuštěný ve vhodném rozpouštědle) dávkován přímo do kapilární kolony mikrostříkačkou opatřenou speciální tenkou jehlou. Tato technika dávkování se používá pro některé stopové analýzy jako alternativa ke klasickému nástřiku provozovanému v režimu s uzavřeným děličem nebo obecně v případech, kdy vzorek obsahuje vysokovroucí látky, u kterých by hrozilo riziko neúplného odpaření vzorku v injektoru SSL, čímž by při analýze došlo k tzv. diskriminaci těžkých podílů. Při diskriminaci by tak byl stanovený obsah vysokovroucích podílů nižší, než je jejich skutečný obsah ve vzorku, protože část vysokovroucích složek by se nedostala do analytické kolony a následně do detektoru. Při použití nástřiku typu COC jsou všechny složky vzorku kvantitativně převedeny do chromatografické kolony a riziko diskriminace těžkých podílů se tak minimalizuje. Objem kapalných vzorků dávkovaných do injektoru typu COC, bývá obvykle v řádu 10 0 µl. Do všech výše zmíněných typů injektorů mohou být vzorky dávkovány ručně nebo s využitím automatickým dávkovače vzorků (autosampler), díky kterému je pak možné uskutečnit velký počet analýz bez lidského zásahu. Objem plynu dávkovaného na analýzu se obvykle pohybuje v rozmezí µl. Pro dávkování plynných vzorků je naprosto dostačující injektor typu SSL v kombinaci s použitím speciální plynotěsné stříkačky. Pro dávkování především plynů může být plynový chromatograf rovněž vybaven speciálním dávkovacím zařízením, které se skládá z vícecestného ventilu a dávkovací smyčky. Plyn určený k analýze prochází ventilem a dávkovací smyčkou o určitém objemu mimo chromatografický systém. V okamžiku startu analýzy je vícecestný ventil ručně nebo elektricky přepnut do polohy, v níž se smyčka naplněná plynem zařadí do proudu mobilní fáze a analyt je tak dopraven do analytické kolony. Výhoda takového dávkovače spočívá v lepší opakovatelnosti dávkovaného množství vzorku, a především pak v možnosti automatizace. Plynové chromatografy vybavené výše zmíněným dávkovacím zařízením se často používají jako on-line analyzátory, které automaticky provádějí opakovanou analýzu plynů v různých provozech. Obecně se v plynové chromatografii uplatňují i jiné typy nástřiků. Příkladem jsou nástřik typu headspace, nástřiky založené na tepelné desorpci (s využitím SPME vlákna nebo systému purge and trap ) nebo speciální pyrolýzní nástřiky. Nástřiky typu headspace (z angl. prostor hlavy) se používají pro dávkování plynné fáze, která je v rovnováze s kapalnou fázi. Vzorek je po určitou dobu uzavřen v termostatované vialce, přičemž postupně dochází k ustavování rovnováhy mezi plynnou a kapalnou fázi. Následně je 36
37 část plynné fáze (nachází se nad fázi kapalnou, proto headspace) odebrána stříkačkou manuálně nebo autosamplerem a nastříknuta do plynového chromatografu. Při headspace analýze se nejčastěji využívají nástřikové členy typu SSL nebo PTV. Lze tak provádět analýzu těkavých látek. Nástřiky založené na tepelné desorpci spočívají v navázání analytu na vhodný sorbent, ze kterého jsou následně uvolněny zahřátím na vysokou teplotu. Příkladem je využití SPME, neboli mikroextrakce na tuhou fázi (viz kap. Chyba! Nenalezen zdroj odkazů.). Při tomto typu analýzy ochází v prvním kroku k navázání těkavého analytu na SPME vlákno, které je následně vloženo do nástřikového členu plynového chromatografu, kde v důsledku zvýšené teploty dochází k desorpci analytu z vlákna. Nejčastěji se k tomuto typu analýzy využívají SSL nebo PTV nástřiky. Zvláštním případem analýzy založené na tepelné desorpci je systém purge and trap. V tomto systému jsou kapalné vzorky umístěné v systému ohraničeném z vrchu a zespodu fritami. Kapalný vzorek je po určitou dobu probubláván inertním plynem, který z něj strhává těkavé složky, které vede na adsorbent. Po určitém čase je probublávání zastaveno a přes adsorbent je za zvýšené teploty veden nosný plyn, přičemž dochází k postupnému uvolnění analytů, které jsou vedeny do SSL nebo PTV nástřikového členu plynového chromatografu. Pyrolýzní nástřiky jsou založené na řízeném tepelném rozkladu polymerních vzorků za nepřístupu vzduchu. Produkty pyrolýzy jsou vyhřívanými cestami vedeny do SSL nebo PTV nástřikového členu plynového chromatografu Kolony Kolony představují v chromatografii prostředí, kde dochází k samotné separaci. V plynové chromatografii se používají dva základní typy kolon, a to kolony náplňové a kapilární. Tyto se vzájemně kromě rozměrů (délka, vnitřní průměr, tloušťka stacionární fáze) liší také separační účinností, s čímž také souvisí jejich různé využití. 1. Náplňové kolony jsou v porovnání s kapilárními kolonami výrazně kratší a mají větší vnitřní průměr. Jejich separační účinnost je výrazně menší než separační účinnost kapilárních kolon (max. do 20 složek). Na druhé straně mají v porovnání s kapilárními kolonami vyšší kapacitu, tedy lze na ně dávkovat větší množství vzorků. Náplňové kolony lze rozdělit na klasické náplňové kolony a mikronáplňové kolony. U obou těchto typů kolon může být stacionární fází tuhý zrnitý adsorbent (GSC, viz kap ) nebo kapalina zakotvená na vhodném nosiči (GLC). Klasické náplňové kolony se vyrábí ze skla, polymerů, nebo kovů. Jejich délka se pohybuje řádově v jednotkách metrů, nejčastěji 2 6 m. Vnitřní průměr se pohybuje v jednotkách milimetrů, nejčastěji 2 3 mm. Mikronáplňové kolony jsou nejčastěji vyrobeny ze skla. Jsou obvykle delší a užší než klasické náplňové kolony, mají vyšší separační účinnost, ale nižší kapacitu. 2. Kapilární kolony se vyrábí hlavně z křemene, který je křehký, a proto bývá potažen vrstvou ochranného polymeru (např. polyimid), který zabraňuje jejich lámání. Kapilární kolony mají 37
38 délku řádově v desítkách metrů, obvykle m. Vnitřní průměr se pohybuje řádově v desetinách milimetrů. Vyznačují se nízkou kapacitou, ale vysokou separační účinností. Tyto kolony umožňují v rámci jedné analýzy rozdělit až stovky složek. Kapilární kolony lze podle způsobu umístění stacionární fáze rozdělit do tří základních skupin, které jsou uvedeny níže. WCOT (z angl. wall coated open tubular): kapalná stacionární fáze je nanesena na vnitřní stěně kolony. Jsou vhodné zejména k analýze kapalných vzorků s nízkým obsahem velmi těkavých složek. Mají vysokou separační schopnost, umožňují provádět detailní analýzu pro směsi typu benzinů obsahující přibližně složek. Nepolární fáze (např. polydimethylsiloxan PDMS) dělí uhlovodíkové směsi na základě bodů varu, pro vzorky obsahující heteroatomy (např. O, S, N, atd.) toto platí pouze omezeně. Polární fáze (např. polyethylenglykol PEG) dělí složky na základě jejich polarity. PLOT (z angl. porous layer open tubular): stacionární fázi je adsorbent zachycen na vnitřní stěně kolony. Jsou vhodné zejména pro analýzu permanentních plynů a velmi těkavých sloučenin. SCOT (z angl. support coated open tubular): kapalná stacionární fáze zakotvena na nosiči zachyceném na vnitřní stěně kolony. Mají ještě vyšší separační schopnost než kolony typu WCOT a lze je tedy použít pro komplexní kapalné směsi s vysokým počtem složek. Běžně používané plynově-chromatografické kolony nelze zahřívat na teploty vyšší než C neboť by mohlo dojít k nevratnému poškození jejich stacionární fáze, případně ochranné polymerní vrstvy. Specifické typy chromatografických kolon, tzv. vysokoteplotní kolony, lze využít k separacím, které probíhají až za teplot C. Mluvíme pak o vysokoteplotní plynové chromatografii (HT-GC, z angl. high temperature gas chromatography). Tato technika se v analytice paliv používá zejména pro analýzu těžších ropných frakcí, viz také kap ) Využití různých typů plynově-chromatografických kolon v analýze paliv V plynové chromatografii se v současné době používají především křemenné a kovové kapilární analytické kolony, nicméně lze se setkat i s klasickými náplňovými a mikronáplňovými kolonami, které se využívají spíše při analýze plynů nebo při aplikaci jiných speciálních metod. Co se týče typu kolon a stacionární fáze, při analýze kapalných paliv se obecně nejvíce používají kapilární kolony s chemicky zakotvenou nepolární stacionární fází WCOT polydimethylsiloxanového typu. Na tomto typu kolon eluují z kolony látky v pořadí podle jejich bodu varu. Rozměry kolony a tloušťka filmu stacionární fáze závisí na konkrétní aplikaci, ale pro představu lze uvést, že délka kapilárních kolon se obvykle pohybuje v rozmezí 5 až 100 m, vnitřní průměr v rozmezí 0,1 až 0,5 mm a tloušťka filmu stacionární fáze 0,1 až 5 µm. Při některých analytických metodách se ale používají i kolony s polární stacionárních jako např. například polyethylenglykol (PEG) či 1,2,3-tris[2-cyanoethoxy]propan (TCPE) nebo dokonce kombinace dvou či více kolon s diametrálně odlišnou polaritou, případně s jiným separačním mechanismem (např. molekulová síta). Přestože plynná paliva jsou z hlediska počtu složek nejjednodušší, pro analýzu plynů (zemní plyn, LPG, topné plyny apod.) nejsou příliš vhodné klasické kapilární kolony se stacionární 38
39 fází typu PDMS, protože na nich dochází k nedokonalému dělení především C4 nenasycených uhlovodíků a jejich různých izomérů. Velmi problematické je pak dělení permanentních plynů, které jsou v některých plynných palivech rovněž obsaženy (N2, O2, CO, CO2, H2). Pro dělení uhlovodíkových složek se proto při analýze plynů používají spíše kapilární kolony typu PLOT se stacionární fází typu modifikované aluminy nebo náplňové kolony plněné molekulovými síty, případně sorbenty s jinou stacionární fází. Při analýze složitějších plynů obsahující jak uhlovodíkové složky, tak permanentní plyny se často využívají složitější vícekolonové chromatografické techniky. Speciální vícekolonové chromatografické techniky se používají při některých analýzách plynů i kapalných paliv. Nejjednodušší vícekolonové techniky spočívají v sériovém spojení dvou kolon s odlišnou stacionární fází. Všechny komponenty vzorku tak procházejí první a následně druhou kolonou, ze které je analyt veden do detektoru. Chromatografický záznam má standardní podobu závislosti velikosti signálu na retenčním čase. Složitější vícekolonové techniky jsou obecně označovány jako metody multidimenzionální plynové chromatografie (MDGC), které spočívají v použití dvou nebo více kolon s odlišnou stacionární fází, přičemž zpravidla pouze část vzorku eluovaná z jedné kolony je následně separována na dalších koloně/kolonách. K tomu slouží systém ventilů, které v přesně stanovený čas přepínají tok mobilní fáze mezi jednotlivými kolonami. Při aplikaci MDGC se často používá zpětný výplach kolony (tzv. backflusch), což je obrácení toku mobilní fáze v koloně. Vybrané látky tak do kolony vstupují i z kolony vystupují na stejném konci. Tímto způsobem se obvykle z kolony vyplachují silně zadržované látky poté, co část slaběji zadržovaných komponent kolonou prošla. I v oblasti analýzy paliv se lze setkat s patrně nejsofistikovanější metodou MDGC, tzv. komprehenzivní plynovou chromatografií označovanou též zkratkou GC GC. Při této technice se používají dvě kolony zpravidla s odlišnou polaritou, přičemž první kolona je v porovnání s druhou poměrně krátká (např. 30 a 0,5 m). Spolu s tzv. modulátorem umístěným mezi kolonami je pak celý vzorek separován na obou kolonách, přičemž chromatografický záznam poskytuje informace o retenčním čase dané komponenty na obou kolonách. Chromatografický záznam je tedy trojrozměrný (na osách x a y jsou retenční časy na první, resp. druhé koloně a na ose z je velikost signálu detektoru), ale pro větší přehlednost bývá často zobrazován jako tzv. contour plot (na osách x a y jsou retenční časy na první, resp. druhé koloně, intenzita signálu detektoru je zobrazena různými barvami) Detektory Ionizační detektory Ionizační detektory představují nejpočetněji zastoupenou skupinu detektorů v plynové chromatografii. Jejich princip spočívá v ionizaci vzorku, při které vznikají ionty, neboli nabité částice, které v elektrickém poli způsobí nárůst elektrického proudu. Podle způsobu, jakým dochází k ionizaci, rozlišujeme tyto typy ionizačních detektorů: plamenově-ionizační detektor (FID), klasický a různé modifikace O-FID, A-FID, NPD, fotoionizační detektor (PID), detektor elektronového záchytu (ECD), 39
40 heliový a argonový detektor. Plamenově-ionizační detektor (FID, z angl. flame-ionization detector) je jednoznačně nejpoužívanější detektor v plynově-chromatografické analýze paliv. Vyplývá to z toho, že kapalná a plynná paliva jsou složena z organických látek, kterými jsou navíc téměř výhradně uhlovodíky. Již z názvu plyne, že k ionizaci dochází v plamenu, tedy dochází k spálení analytu v kyslíkovovodíkovém plamenu za vzniku iontů. Tento detektor z hořlavých palivových složek nedetekuje pouze CO, H2 a H2S. Pro organické látky je FID detektor univerzální, protože je dostatečně citlivý a vykazuje lineární odezvu v širokém rozsahu koncentrací. Hmotnostní odezva FID detektoru na různé uhlovodíky je navíc velice podobná. V závislosti na zvolené referenční látce (s výjimkou methanu) se od sebe obvykle odezvy uhlovodíků neliší o více než 5 %, v extrémních případech nepřesahují relativní rozdíly 12 %. Toho lze s výhodou využít při kvantifikaci obsahu jednotlivých uhlovodíků nebo skupin uhlovodíků. Za předpokladu stejné odezvy všech uhlovodíků lze obsah (% hm.) libovolné látky v čistě uhlovodíkové směsi vypočítat pouze na základě procentuálního podílu plochy píku zvolené látky a součtu plochy všech píků (% plochy), aniž by bylo nutné dělat jakoukoliv kalibraci. S předpokladem stejné odezvy všech uhlovodíků pracují například všechny standardizované metody simulované destilace. Tam kde je třeba stanovení zpřesnit se pro výpočet obsahu používají buď tzv. metoda vnitřní normalizace pracující s relativními odezvovými faktory (poměr odezvy dané složky a referenční látky) nebo standardní kalibrační metoda (absolutní kalibrace, metoda vnitřního standardu apod.). Metodu vnitřní normalizace nebo nějakou standardní kalibrační metodu je třeba použít vždy, když vzorek obsahuje jiné látky než uhlovodíky (např. automobilové benzíny s obsahem kyslíkatých látek nebo motorové nafty s obsahem bionafty). Kyslíkový FID detektor (O-FID, z angl. oxygen flame ionization detector) je selektivním detektorem pro stanovení kyslíkatých sloučenin. Před samotným FID detektorem jsou zařazený krakovací reaktor, který převádí analyty obsahující kyslík na CO, který je následně v methanizéru převeden na methan. Vzniklý methan je následně stanoven FID detektorem. Termoionizační detektor se solí alkalického kovu (A-FID, z angl. alkaline flame ionization detector) představuje modifikovaný FID detektor. V literatuře se označuje také TID (z angl. thermionic ionization detector). Princip se však od klasického FID detektoru a jiných ionizačních detektorů liší. AFID je selektivním detektorem pro organické sloučeniny obsahující fosfor, arsen, bor, síru, halogenidy, atd. Do blízkosti hořáku je vložen prstenec s vhodnou solí alkalického kovu. Přítomnost analytu v detektoru má za následek změnu v ionizaci alkalického kovu, což se projeví zvýšením anebo snížením ionizačního proudu v závislosti na povaze analytu. Dusíkovo-fosforový detektor (NPD, z angl. nitrogen-phosphorus detector) pracuje na stejném principu jako A-FID. Prstenec obsahuje sůl rubidia nebo cesia, které zabezpečují selektivitu tohoto detektoru vůči dusíku a fosforu. NPD detektor se vyznačuje velmi vysokou citlivostí. Fotoionizační detektor (PID, z angl. photoionization detector) je detektor selektivní pro stanovení aromátů, olefinů a sloučeniny obsahující síru nebo fosfor. Již z názvu plyne, že k ionizaci 40
41 dochází v důsledku ozáření vzorku UV zářením. Vlnová délka záření ovlivňuje selektivitu stanovení. Jedná se o nedestruktivní detektor vhodný pro polní podmínky. Detektor elektronového záchytu (ECD, z angl. electron capture detector) je selektivním detektorem pro halogensloučeniny, nitráty a konjugované karbonyly. Základem je -zářič 63 Ni, který je zdrojem primárních elektronů ( -částic), které ionizují molekuly nosného plynu, čím produkují sekundární elektrony. Tento detektor poskytuje odezvu na sloučeniny, které jsou schopny zachycovat volné elektrony a tvořit anionty. Signál detektoru se tvoří v důsledku snížení počtu volných elektronů, což vede ke snížení proudového signálu. ECD detektor má velmi vysoké nároky na čistotu nosného plynu a vyznačuje se velmi vysokou citlivostí. Heliový a argonový detektor představují dva různé detektory, které ale pracují na stejném principu. V obou případech se jedná o ionizační detektory. K ionizaci vzorku dochází v důsledku interakce s metastabilními atomy hélia (argonu). Metastabilního stavu jednoatomového nosného plynu je dosaženo emisí elektronů z radioaktivního -zářiče (tritium, 63 Ni) za vysokého napětí. Tyto detektory jsou univerzální, lze je použít pro analýzu permanentních plynů. Citlivost je podobná jako u FID detektoru. Nároky na čistotu nosného plynu jsou ještě vyšší než pro ECD detektor Další detektory Tepelně-vodivostní detektor (TCD, z angl. thermal conductivity detector) je poměrně široce využívaným detektorem v analýze paliv. Podobně jako FID je univerzální, avšak vůči organickým látkám vykazuje obecně nižší citlivost. Odezva detektoru je navíc pro různé látky odlišná, takže pro kvantifikaci je třeba provádět kalibraci pro každou stanovovanou složku. Použití TCD detektoru je však v podstatě nezbytné při analýze plynných paliv obsahujících permanentní plyny, protože ty nelze detekovat FID detektorem. Při analýze takového typu plynných paliv se lze často setkat s kombinací obou výše zmíněných detektorů, přičemž FID detektor se používá pro detekci uhlovodíků a TCD detektor pro detekci permanentních plynů. Hmotnostní detektor se používá ve spojení s plynovou i kapalinovou chromatografií. Blíže bude charakterizován v kap. 5. Sírný chemiluminiscenční detektor (SCD, z angl. sulfur chemiluminescence detector) Dusíkový chemiluminiscenční detektor (NCD, z angl. nitrogen chemiluminescence detector) Plamenofotometrický (plamenoemisní) detektor (FPD, z angl. flame photometric detector) je selektivním detektorem pro sloučeniny obsahující síru a fosfor. Analyt je spálen v nízkoteplotním plamenu bohatém na vodík. Sloučeniny obsahující síru emitují záření s vlnovou délkou 394 nm. Sloučeniny obsahující fosfor emitují záření s vlnovou délkou 526 nm. Tento detektor nedokáže současné detekovat síru a fosfor a pro síru má nelineární odezvu. FPD detektor s pulzním plamenem (PFPD) má přibližně desetkrát vyšší citlivost než běžný FPD Detektory pro plynovou chromatografii shrnutí Nejpoužívanějším detektorem v plynově-chromatografické analýze paliv je jednoznačně pro uhlovodíky univerzální FID detektor. Mezi další často využívané detektory patří hmotnostní 41
42 detektor a TCD detektor. Ostatní typy detektorů se pro rutinní analýzu paliv používají již méně často, lze se s nimi setkat při stanovení stopových koncentrací vybraných látek s použitím citlivých selektivních detektorů nebo v oblasti výzkumu (selektivní detektory či MS detektor). Příkladem může být stanovení sirných sloučenin v zemním plynu s využitím selektivních sirných detektorů (např. FPD, SCD či AED) nebo například stanovení látek typu PCB v uhlovodíkových matricích. Spojení plynové chromatografie s hmotnostní spektrometrií (GC-MS) se v oblasti paliv většinou využívá především pro bližší identifikaci neznámých látek při speciálních analýzách nestandardních vzorků, avšak i přes poněkud obtížnější kvantifikaci se techniky GC-MS používají v současné době i pro rutinní stanovení některých látek. Speciální selektivní detektory pak nacházejí uplatnění využití především při stanovení stopových koncentrací organických látek, jejíž stanovení není proveditelné s využitím univerzálních detektorů více či méně z důvodu jejich nižší citlivosti, ale především z důvodu nemožnosti identifikace a integrace píku hledané komponenty koelující s jinými organickými látkami. Přehled nejpoužívanějších selektivních detektorů je uveden tab Tab. 3.2: Přehled nejpoužívanějších selektivních detektorů Zkratka Název Selektivita Poznámka ECD detektor elektronového záchytu halogenované látky, nitráty, konjugované karbonyly O-FID selektivní kyslíkový FID detektor látky obsahující kyslík (P)FPD NPD SCD NCD AED (pulsní) plamenový fotometrický detektor dusíkovo-fosforový detektor sirný chemiluminiscenční detektor dusíkový chemiluminiscenční detektor atomový emisní detektor látky obsahující síru/fosfor látky obsahující dusík a fosfor látky obsahující síru látky obsahující dusík prvkově selektivní (C, S, N, Cl, ) Aplikace plynové chromatografie v analýze paliv obsahuje radioaktivní izotop 63 Ni k vlastní detekci složí FID detektor detekující metan vznikající metanizací CO nelineární odezva na sirné látky termionický specifický detektor (TSD) extrémně citlivý a selektivní extrémně citlivý a selektivní atomizace a excitace analytu v plazmě Plynová chromatografie má v analýze paliv nezastupitelnou úlohu. U jednodušších paliv je možné plynovou chromatografií provádět detailní individuální analýzu všech zastoupených komponent, jejímž výsledkem jsou kvalitativní i kvantitativní informace o všech zastoupených složkách. S rostoucím počtem složek v palivu se stává stanovení kompletního individuálního složení stále obtížnější až neproveditelné. Problematiku stanovení individuálního složení paliv lze dobře ilustrovat na příkladu klasických fosilních paliv. Pomineme-li minoritní zastoupeních některých organických látek obsahujících především síru, dusík, kyslík a permanentní plyny doprovázející zemní plyn, skládají se paliva vyráběná ze zemního plynu a ropy pouze z uhlovodíků, tedy organických látek obsahujících ve svých molekulách pouze atomy uhlíku a vodíku. Z uhlovodíků obsahuje zemní plyn především nejjednodušší uhlovodík, tedy metan (obvykle 42
43 více než 90 obj. %), v menším množství pak nasycené uhlovodíky se dvěma až pěti uhlíkovými atomy v molekule. Obsah vyšších uhlovodíků je zcela zanedbatelný. Nejznámějším nejjednodušším palivem vyráběným z ropy je tzv. LPG (z angl. liqufied petroleum gas) označovaný též příznačně jako propan-butan. Posledně uvedený název do jisté míry prozrazuje složení tohoto paliva. LPG však neobsahuje pouze propan a butany (i-butan a n-butan), ale obvykle i menší množství etanu a C5 uhlovodíků. LPG navíc obsahuje i nenasycené uhlovodíky (propen, buteny, příp. butadieny), takže reálně se dostáváme přibližně na 15 uhlovodíkových složek. V automobilovém benzínu lze již identifikovat přibližně 300 uhlovodíků a počet uhlovodíků přítomných v motorové naftě se již pohybuje v řádu tisíců až milionů různých sloučenin. Celkový počet uhlovodíků přítomných v palivu souvisí s jeho destilačním rozmezím, resp. s rozmezím počtu atomů uhlíku přítomných v daném palivu. Budeme-li uvažovat pouze nasycené alifatické uhlovodíky, existuje pouze jeden jejich zástupce od uhlovodíků C1 až C3. Ve skupině C4 alkanů jsou dva (i-butan a n-butan), ve skupině C5 tři, ve skupině C6 pět a dále počet možných izomérů narůstá geometrickou řadou. Na obr. 3.1 je pro ilustraci uveden přibližný rozsah počtu atomů uhlíku a počet možných izomérů alifatických alkanů v různých ropných produktech. Každý možný izomer sice nemusí být nutně v daném palivu zastoupen, avšak na druhou stranu obsahují ropné produkty kromě alifatických nasycených uhlovodíků i uhlovodíky alkeny, cykloalkany, aromáty a jejich alkylované homology. Počet možných izomerů těchto uhlovodíků přitom s rostoucím počtem atomů uhlíku v molekule pochopitelně rovněž roste. V praxi to znamená, že stanovení zastoupení všech komponent je možné pouze u zemního plynu, LPG a benzínu (automobilový, letecký, technický, apod.) S využitím moderních chromatografických metod je možné identifikovat a stanovit i většinu látek přítomných v petrolejových frakcích (např. letecký petrolej), případně i v motorových naftách, avšak taková analýza je náročná a z praktického hlediska není příliš účelná. Kromě stanovení individuálního složení (všechny složky) se používá plynová chromatografie i pro stanovení pouze vybraných látek nebo vybraných skupin látek. V některých případech se přitom rozšiřuje oblast zájmu i na jiné organické látky než uhlovodíky, ať už se jedná o látky do uhlovodíkových paliv účelově přidávané (např. bioetanol nebo bionafta), či látky vyskytující se v palivu přirozeně ve stopovém množství. Příkladem může být například stanovení obsahu benzenu, aromatických uhlovodíků či kyslíkatých látek v benzínu, stanovení obsahu bionafty v motorové naftě nebo např. stanovení sirných látek v zemním plynu. 43
44 1.E+18 1.E+16 těžké topné a mazací oleje 1.E+14 Počet izomérů uhlovodíků 1.E+12 1.E+10 1.E+08 1.E+06 1.E+04 LPG benzin petrolej nafta 1.E+02 1.E+00 Obr. 3.1: Počet atomů uhlíku Přibližný rozsah počtu atomů uhlíku a počet možných izomérů alifatických alkanů v různých ropných produktech Bez ohledu na destilační rozmezí, resp. počet komponent přítomných v palivu má plynová chromatografie univerzální využití jako metoda tzv. otisku prstu (fingerprint), která spočívá pouze v získání charakteristického chromatografického záznamu vzorku, který se pak porovnává se záznamy referenčních materiálů získaných při stejných chromatografických podmínkách. Jedná se v podstatě o nejjednodušší aplikaci plynové chromatografie, protože chromatografický záznam získáme při jakékoliv chromatografické analýze. Přestože informace o analyzovaném vzorku mohou být v některých případech značně omezené, je metoda otisku prstu často užitečná pro rychlé posouzení typu analyzovaného materiálu (viz obr. 3.2) či pro určení identity vzorku s referenčním materiálem. Podobným způsobem se využívá metoda otisku prstu i u jiných analytických metod (např. v infračervené spektrometrii). Podobné univerzální využití má plynová chromatografie při stanovení destilační charakteristiky paliv a dalších ropných produktů. Taková aplikace plynové chromatografie se nazývá simulovaná destilace. Simulovaná destilace má svá specifika i jistá omezení, nicméně hlavním rozdílem oproti ostatním chromatografickým metodám je skutečnost, že simulovanou destilací nezískáváme primárně informace o přítomnosti a obsahu konkrétních látek, nýbrž pouze informaci o destilačním křivce analyzovaného vzorku (tabelovaná nebo grafická závislost teploty varu na předestilovaném množství). 44

45 letecký petrolej těžký benzín lehký topný olej Signál plynový olej (nafta) Retenční čas (min) Obr. 3.2: Porovnání chromatogramů různých ropných produktů Analýza plynů Plynově-chromatografická analýza uhlovodíkových plynů typu LPG je poměrně jednoduchá. Ať už se jedná o čistý propan, či butan nebo směs plynů určenou pro pohon motorů či topné účely, obsahují tato paliva uhlovodíkové plyny s převládající zastoupením C3 a C4 uhlovodíků. Kromě toho mohou obsahovat i malé množství metanu, C2 a C5 uhlovodíků, případně stopy vyšších uhlovodíků (> C5). Metody pro analýzu LPG jsou standardizované, jako příklad lze uvést normu ČSN EN Vzorek LPG se do plynového chromatografu dávkuje v kapalném nebo plynném stavu pomocí speciálního dávkovacího zařízení, avšak pro dávkování plynného vzorku je možné principiálně použít standardní SSL nástřik v kombinaci s použitím plynotěsné stříkačky pro vlastní dávkování vzorku. Před vlastní analýzou plynného vzorku je nejdříve třeba provést jeho řízené zplynění (LPG je skladován v tlakových nádobách v kapalném stavu). Pro detekci se používá přednostně FID detektor, ale použít lze i detektor typu TCD. Obsah všech složek vzorku se vypočítá s využitím vnitřní normalizace, tzn. na základě znalosti ploch chromatografických píků a relativních faktorů odezvy jednotlivých složek, jak je uvedeno v rovnici w i = F i A i 100 (3.26) F i A i kde wi je obsah složky (% hm.) ve směsi, Fi je faktor odezvy i-té složky vzorku a Ai plocha píku příslušné složky. Referenční látka s relativním odezvovým faktorem rovným 1,00 je v tomto případě n-butan. Analytická kolona pro separaci jednotlivých složek vzorku může být buď náplňová (různé polární fáze zakotvené na Chromosorbu P či jiném vhodném nosiči), nebo kapilární (kolony typu 45
Separační metody Historie: Rozvoj separačních metod od minulého století Postavení separačních metod v rámci analytické chemie Význam chromatografie a
 Úvod do separačních metod pro analýzu léčiv Příprava předmětu byla podpořena projektem OPP č. CZ..7/3..00/3353 Separační metody Historie: Rozvoj separačních metod od minulého století Postavení separačních
Úvod do separačních metod pro analýzu léčiv Příprava předmětu byla podpořena projektem OPP č. CZ..7/3..00/3353 Separační metody Historie: Rozvoj separačních metod od minulého století Postavení separačních
EXTRAKČNÍ METODY. Studijní materiál. 1. Obecná charakteristika extrakce. 2. Extrakce kapalina/kapalina LLE. 3. Alkalická hydrolýza
 Studijní materiál EXTRAKČNÍ METODY 1. Obecná charakteristika extrakce 2. Extrakce kapalina/kapalina LLE 3. Alkalická hydrolýza 4. Soxhletova extrakce 5. Extrakce za zvýšené teploty a tlaku PLE, ASE, PSE
Studijní materiál EXTRAKČNÍ METODY 1. Obecná charakteristika extrakce 2. Extrakce kapalina/kapalina LLE 3. Alkalická hydrolýza 4. Soxhletova extrakce 5. Extrakce za zvýšené teploty a tlaku PLE, ASE, PSE
METODY ČIŠTĚNÍ ORGANICKÝCH LÁTEK
 METODY ČIŠTĚNÍ ORGANICKÝCH LÁTEK Chemické sloučeniny se připravují z jiných chemických sloučenin. Tento děj se nazývá chemická reakce, kdy z výchozích látek (reaktantů) vznikají nové látky (produkty).
METODY ČIŠTĚNÍ ORGANICKÝCH LÁTEK Chemické sloučeniny se připravují z jiných chemických sloučenin. Tento děj se nazývá chemická reakce, kdy z výchozích látek (reaktantů) vznikají nové látky (produkty).
Destilace
 Výpočtový ý seminář z Procesního inženýrství podzim 2007 Destilace 18.9.2008 1 Tématické okruhy destilace - základní pojmy rovnováha kapalina - pára jednostupňová destilace rektifikace 18.9.2008 2 Destilace
Výpočtový ý seminář z Procesního inženýrství podzim 2007 Destilace 18.9.2008 1 Tématické okruhy destilace - základní pojmy rovnováha kapalina - pára jednostupňová destilace rektifikace 18.9.2008 2 Destilace
Studijní materiál. Úvod do problematiky extrakčních metod. Vypracoval: RNDr. Ivana Borkovcová, Ph.D.
 Studijní materiál Úvod do problematiky extrakčních metod Vypracoval: RNDr. Ivana Borkovcová, Ph.D. Úvod do problematiky extrakčních metod Definice, co je to extrakce separační proces v kontaktu jsou dvě
Studijní materiál Úvod do problematiky extrakčních metod Vypracoval: RNDr. Ivana Borkovcová, Ph.D. Úvod do problematiky extrakčních metod Definice, co je to extrakce separační proces v kontaktu jsou dvě
DĚLÍCÍ METODY. Autor: Mgr. Stanislava Bubíková. Datum (období) tvorby: 28. 5. 2012. Ročník: osmý. Vzdělávací oblast: Člověk a příroda / Chemie / Směsi
 tvorby: 28. 5. 2012. Ročník: osmý. Vzdělávací oblast: Člověk a příroda / Chemie / Směsi") Autor: Mgr. Stanislava Bubíková DĚLÍCÍ METODY Datum (období) tvorby: 28. 5. 2012 Ročník: osmý Vzdělávací oblast: Člověk a příroda / Chemie / Směsi 1 Anotace: Žáci se seznámí s nejčastěji používanými separačními
Autor: Mgr. Stanislava Bubíková DĚLÍCÍ METODY Datum (období) tvorby: 28. 5. 2012 Ročník: osmý Vzdělávací oblast: Člověk a příroda / Chemie / Směsi 1 Anotace: Žáci se seznámí s nejčastěji používanými separačními
Teorie chromatografie - I
 Teorie chromatografie - I Veronika R. Meyer Practical High-Performance Liquid Chromatography, Wiley, 2010 http://onlinelibrary.wiley.com/book/10.1002/9780470688427 Příprava předmětu byla podpořena projektem
Teorie chromatografie - I Veronika R. Meyer Practical High-Performance Liquid Chromatography, Wiley, 2010 http://onlinelibrary.wiley.com/book/10.1002/9780470688427 Příprava předmětu byla podpořena projektem
Inovace bakalářského a navazujícího magisterského studijního programu v oboru Bezpečnost a kvalita potravin (reg. č. CZ.1.07/2.2.00/28.
 Inovace bakalářského a navazujícího magisterského studijního programu v oboru Bezpečnost a kvalita potravin (reg. č. CZ.1.07/2.2.00/28.0287) EXTRAKČNÍ METODY Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana
Inovace bakalářského a navazujícího magisterského studijního programu v oboru Bezpečnost a kvalita potravin (reg. č. CZ.1.07/2.2.00/28.0287) EXTRAKČNÍ METODY Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana
Stanovení křivky rozpustnosti fenol-voda. 3. laboratorní cvičení
 Stanovení křivky rozpustnosti fenol-voda 3. laboratorní cvičení Mgr. Sylvie Pavloková Letní semestr 2016/2017 Cíl pochopení základních principů fázové rovnováhy heterogenních soustav základní principy
Stanovení křivky rozpustnosti fenol-voda 3. laboratorní cvičení Mgr. Sylvie Pavloková Letní semestr 2016/2017 Cíl pochopení základních principů fázové rovnováhy heterogenních soustav základní principy
Gelová permeační chromatografie
 Gelová permeační chromatografie (Gel Permeation Chromatography - GPC) - separační a čisticí metoda - umožňuje separaci skupin sloučenin s podobnou molekulovou hmotností (frakcionace) - analyty jsou po
Gelová permeační chromatografie (Gel Permeation Chromatography - GPC) - separační a čisticí metoda - umožňuje separaci skupin sloučenin s podobnou molekulovou hmotností (frakcionace) - analyty jsou po
Superkritická fluidní extrakce (SFE) Superkritická fluidní extrakce
 Superkritická fluidní extrakce") Superkritická fluidní extrakce (zkráceně SFE, z angl. Supercritical Fluid Extraction) = extrakce, kde extrakčním činidlem je tekutina v superkritickém stavu, tzv. superkritická (nadkritická) tekutina (zkráceně
Superkritická fluidní extrakce (zkráceně SFE, z angl. Supercritical Fluid Extraction) = extrakce, kde extrakčním činidlem je tekutina v superkritickém stavu, tzv. superkritická (nadkritická) tekutina (zkráceně
Fázové heterogenní rovnováhy Fáze = homogenní část soustavy, oddělná fyzickým rozhraním, na rozhraní se vlastnosti mění skokem
 Fázové heterogenní rovnováhy Fáze = homogenní část soustavy, oddělná fyzickým rozhraním, na rozhraní se vlastnosti mění skokem Rovnováha Tepelná - T všude stejná Mechanická - p všude stejný Chemická -
Fázové heterogenní rovnováhy Fáze = homogenní část soustavy, oddělná fyzickým rozhraním, na rozhraní se vlastnosti mění skokem Rovnováha Tepelná - T všude stejná Mechanická - p všude stejný Chemická -
Fázové rovnováhy dvousložkové soustavy kapalina-kapalina
 Fázové rovnováhy dvousložkové soustavy kapalina-kapalina A) Neomezeně mísitelné kapaliny Za situace, kdy se v dvousložkové soustavě vyskytuje jediná kapalná fáze (neomezená mísitelnost obou kapalin), pak
Fázové rovnováhy dvousložkové soustavy kapalina-kapalina A) Neomezeně mísitelné kapaliny Za situace, kdy se v dvousložkové soustavě vyskytuje jediná kapalná fáze (neomezená mísitelnost obou kapalin), pak
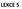 LEKCE 5 Krystalizace separace složek homogenních směsí - krystalizace druhy krystalizace: volná krystalizace rušená krystalizace frakční krystalizace krystalizace změnou složení rozpouštědla vykrývání
LEKCE 5 Krystalizace separace složek homogenních směsí - krystalizace druhy krystalizace: volná krystalizace rušená krystalizace frakční krystalizace krystalizace změnou složení rozpouštědla vykrývání
SPE je metoda vhodná pro rychlou přípravu vzorků, která užívá
 Extrakce na pevné fázi (SPE) Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Extrakce na pevné fázi (SPE) (Solid Phase Extraction) SPE je metoda vhodná pro rychlou přípravu vzorků,
Extrakce na pevné fázi (SPE) Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Extrakce na pevné fázi (SPE) (Solid Phase Extraction) SPE je metoda vhodná pro rychlou přípravu vzorků,
Separační metody v analytické chemii. Plynová chromatografie (GC) - princip
 - princip") Plynová chromatografie (GC) - princip Plynová chromatografie (Gas chromatography, zkratka GC) je typ separační metody, kdy se od sebe oddělují složky obsažené ve vzorku a které mohou být převedeny do plynné
Plynová chromatografie (GC) - princip Plynová chromatografie (Gas chromatography, zkratka GC) je typ separační metody, kdy se od sebe oddělují složky obsažené ve vzorku a které mohou být převedeny do plynné
Laboratoř oboru. Rektifikace. Ústav organické technologie (111) Vedoucí práce: Ing. Tomáš Sommer Umístění práce: budova A, místnost S31
 Vedoucí práce: Ing. Tomáš Sommer Umístění práce: budova A, místnost S31") Laboratoř oboru Ústav organické technologie (111) I Rektifikace Vedoucí práce: Ing. Tomáš Sommer Umístění práce: budova A, místnost S31 1. Úvod Destilace a rektifikace patří mezi nejčastěji používané procesy
Laboratoř oboru Ústav organické technologie (111) I Rektifikace Vedoucí práce: Ing. Tomáš Sommer Umístění práce: budova A, místnost S31 1. Úvod Destilace a rektifikace patří mezi nejčastěji používané procesy
Analýza kofeinu v kávě pomocí kapalinové chromatografie
 Analýza kofeinu v kávě pomocí kapalinové chromatografie Kofein (obr.1) se jako přírodní alkaloid vyskytuje v mnoha rostlinách (např. fazolích, kakaových bobech, černém čaji apod.) avšak nejvíce je spojován
Analýza kofeinu v kávě pomocí kapalinové chromatografie Kofein (obr.1) se jako přírodní alkaloid vyskytuje v mnoha rostlinách (např. fazolích, kakaových bobech, černém čaji apod.) avšak nejvíce je spojován
SIMULOVANÁ A VAKUOVÁ DESTILACE
 VYSOKÁ ŠKOLA CHEMICKO-TECHNOLOGICKÁ V PRAZE Fakulta technologie ochrany prostředí Ústav technologie ropy a alternativních paliv SIMULOVANÁ A VAKUOVÁ DESTILACE Laboratorní cvičení ÚVOD Simulovaná destilace
VYSOKÁ ŠKOLA CHEMICKO-TECHNOLOGICKÁ V PRAZE Fakulta technologie ochrany prostředí Ústav technologie ropy a alternativních paliv SIMULOVANÁ A VAKUOVÁ DESTILACE Laboratorní cvičení ÚVOD Simulovaná destilace
PLYNOVÁ CHROMATOGRAFIE (GC)
") PLYNOVÁ CHROMATOGRAFIE (GC) Dělení látek mezi stacionární a mobilní fázi na základě rozdílů v těkavosti a struktuře (separované látky vykazují rozdílnou chromatografickou afinitu) Metoda vhodná pro látky:
PLYNOVÁ CHROMATOGRAFIE (GC) Dělení látek mezi stacionární a mobilní fázi na základě rozdílů v těkavosti a struktuře (separované látky vykazují rozdílnou chromatografickou afinitu) Metoda vhodná pro látky:
ROLE SEPARAČNÍCH METOD
 ROLE SEPARAČNÍCH METOD Redukce nežádoucích složek - ruší analýzu, poškozují přístroj Rozdělení - frakcionace vzorku podle zvolené charakteristiky Cílená analýza - vysoce selektivní postup Necílená analýza
ROLE SEPARAČNÍCH METOD Redukce nežádoucích složek - ruší analýzu, poškozují přístroj Rozdělení - frakcionace vzorku podle zvolené charakteristiky Cílená analýza - vysoce selektivní postup Necílená analýza
Sada 7 Název souboru Ročník Předmět Formát Název výukového materiálu Anotace
 Sada 7 Název souboru Ročník Předmět Formát Název výukového materiálu Anotace VY_52_INOVACE_737 8. Chemie notebook Směsi Materiál slouží k vyvození a objasnění pojmů (klíčová slova - chemická látka, směs,
Sada 7 Název souboru Ročník Předmět Formát Název výukového materiálu Anotace VY_52_INOVACE_737 8. Chemie notebook Směsi Materiál slouží k vyvození a objasnění pojmů (klíčová slova - chemická látka, směs,
Rovnováha Tepelná - T všude stejná
 Fázové heterogenní rovnováhy Fáze = homogenní část soustavy, oddělná fyzickým rozhraním, na rozhraní se vlastnosti mění skokem Rovnováha Tepelná - T všude stejná Mechanická - p všude stejný Chemická -
Fázové heterogenní rovnováhy Fáze = homogenní část soustavy, oddělná fyzickým rozhraním, na rozhraní se vlastnosti mění skokem Rovnováha Tepelná - T všude stejná Mechanická - p všude stejný Chemická -
Transportní jevy v plynech Reálné plyny Fázové přechody Kapaliny
 Transportní jevy v plynech Reálné plyny Fázové přechody Kapaliny Hustota toku Zatím jsme studovali pouze soustavy, které byly v rovnovážném stavu není-li soustava v silovém poli, je hustota částic stejná
Transportní jevy v plynech Reálné plyny Fázové přechody Kapaliny Hustota toku Zatím jsme studovali pouze soustavy, které byly v rovnovážném stavu není-li soustava v silovém poli, je hustota částic stejná
CHROMATOGRAFIE ÚVOD Společný rys působením nemísících fází: jedna fáze je nepohyblivá (stacionární), druhá pohyblivá (mobilní).
, druhá pohyblivá (mobilní).") CHROMATOGRAFIE ÚOD Existují různé chromatografické metody, viz rozdělení metod níže. Společný rys chromatografických dělení: vzorek jako směs látek - složek se dělí na jednotlivé složky působením dvou
CHROMATOGRAFIE ÚOD Existují různé chromatografické metody, viz rozdělení metod níže. Společný rys chromatografických dělení: vzorek jako směs látek - složek se dělí na jednotlivé složky působením dvou
Gymnázium a Střední odborná škola, Rokycany, Mládežníků 1115
 Gymnázium a Střední odborná škola, Rokycany, Mládežníků 1115 Číslo projektu: CZ.1.07/1.5.00/34.0410 Číslo šablony: III/2 Inovace a zkvalitněni výuky prostřednictvím ICT. Název materiálu: Zpracování ropy
Gymnázium a Střední odborná škola, Rokycany, Mládežníků 1115 Číslo projektu: CZ.1.07/1.5.00/34.0410 Číslo šablony: III/2 Inovace a zkvalitněni výuky prostřednictvím ICT. Název materiálu: Zpracování ropy
EXTRAKČNÍ METODY používané pro stanovení lipofilních a hydrofilních látek. Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana Borkovcová, Ph.D.
 EXTRAKČNÍ METODY používané pro stanovení lipofilních a hydrofilních látek Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana Borkovcová, Ph.D. EXTRAKČNÍ METODY Úvod rozdělení látek podle polarity extrakce lipofilních
EXTRAKČNÍ METODY používané pro stanovení lipofilních a hydrofilních látek Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana Borkovcová, Ph.D. EXTRAKČNÍ METODY Úvod rozdělení látek podle polarity extrakce lipofilních
zpracování těžkých frakcí na motorová paliva (mazut i vakuový zbytek)
") Ropa štěpné procesy zpracování těžkých frakcí na motorová paliva (mazut i vakuový zbytek) typy štěpných procesů: - termické krakování - katalytické krakování - hydrogenační krakování (hydrokrakování) podmínky
Ropa štěpné procesy zpracování těžkých frakcí na motorová paliva (mazut i vakuový zbytek) typy štěpných procesů: - termické krakování - katalytické krakování - hydrogenační krakování (hydrokrakování) podmínky
Separační metoda. Fázový diagram
 Separační metody využívají se k izolování (separaci) dokazované nebo stanovované složky z analyzované směsi a k odstranění rušivých (interferujících) komponent analyzovaného roztoku (účel získání čistých
Separační metody využívají se k izolování (separaci) dokazované nebo stanovované složky z analyzované směsi a k odstranění rušivých (interferujících) komponent analyzovaného roztoku (účel získání čistých
Vysokoúčinná kapalinová chromatografie
 Vysokoúčinná kapalinová chromatografie HPLC High Performance Liquid Chromatography Vysokoúčinná...X... Vysoceúčinná kapalinová chromatografie RRLC Rapid Resolution Liquid Chromatography Rychle rozlišovací
Vysokoúčinná kapalinová chromatografie HPLC High Performance Liquid Chromatography Vysokoúčinná...X... Vysoceúčinná kapalinová chromatografie RRLC Rapid Resolution Liquid Chromatography Rychle rozlišovací
Rozpustnost Rozpustnost neelektrolytů
 Rozpustnost Podobné se rozpouští v podobném látky jejichž molekuly na sebe působí podobnými mezimolekulárními silami budou pravděpodobně navzájem rozpustné. Př.: nepolární látky jsou rozpustné v nepolárních
Rozpustnost Podobné se rozpouští v podobném látky jejichž molekuly na sebe působí podobnými mezimolekulárními silami budou pravděpodobně navzájem rozpustné. Př.: nepolární látky jsou rozpustné v nepolárních
Příprava materiálu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253
 Příprava materiálu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Část 9 Adsorpční chromatografie: Chromatografie v normálním módu Tento chromatografický mód je vysvětlen na silikagelu jako nejdůležitějším
Příprava materiálu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Část 9 Adsorpční chromatografie: Chromatografie v normálním módu Tento chromatografický mód je vysvětlen na silikagelu jako nejdůležitějším
MODELY SORPCE VOC V ZEMINÁCH VS. METODY STATICKÉ HEAD-SPACE A KAPALINOVÉ EXTRAKCE
 VYSOKÁ ŠKOLA CEICKO-TECNOLOICKÁ V PRAZE Fakulta technologie ochrany prostředí ODELY SORPCE VOC V ZEINÁC VS. ETODY STATICKÉ EAD-SPACE A KAPALINOVÉ EXTRAKCE Veronika Kučerová Doc. Ing. Josef Janků, CSc.
VYSOKÁ ŠKOLA CEICKO-TECNOLOICKÁ V PRAZE Fakulta technologie ochrany prostředí ODELY SORPCE VOC V ZEINÁC VS. ETODY STATICKÉ EAD-SPACE A KAPALINOVÉ EXTRAKCE Veronika Kučerová Doc. Ing. Josef Janků, CSc.
isolace analytu oddělení analytu od matrice (přečištění) zakoncentrování analytu stanovení analytu (analytů) ve vícesložkové směsi
 zakoncentrování analytu stanovení analytu (analytů) ve vícesložkové směsi") SEPARAČNÍ METODY Využití separačních metod isolace analytu oddělení analytu od matrice (přečištění) zakoncentrování analytu stanovení analytu (analytů) ve vícesložkové směsi Druhy separačních metod Srážení
SEPARAČNÍ METODY Využití separačních metod isolace analytu oddělení analytu od matrice (přečištění) zakoncentrování analytu stanovení analytu (analytů) ve vícesložkové směsi Druhy separačních metod Srážení
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ. Stanovení těkavých látek
 LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení těkavých látek (metoda: plynová chromatografie s hmotnostně spektrometrickým detektorem) Garant úlohy: doc. Ing. Jana Pulkrabová, Ph.D. 1 OBSAH
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení těkavých látek (metoda: plynová chromatografie s hmotnostně spektrometrickým detektorem) Garant úlohy: doc. Ing. Jana Pulkrabová, Ph.D. 1 OBSAH
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU DEKOCHINÁTU METODOU HPLC
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU DEKOCHINÁTU METODOU HPLC 1 Rozsah a účel Tato metoda specifikuje podmínky pro stanovení dekochinátu metodou vysokoúčinné kapalinové chromatografie
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU DEKOCHINÁTU METODOU HPLC 1 Rozsah a účel Tato metoda specifikuje podmínky pro stanovení dekochinátu metodou vysokoúčinné kapalinové chromatografie
Zpracování ropy doc. Ing. Josef Blažek, CSc. 6. přednáška
 ODBORNÉ VZDĚLÁVÁNÍ ÚŘEDNÍKŮ PRO VÝKON STÁTNÍ SPRÁVY OCHRANY OVZDUŠÍ V ČESKÉ REPUBLICE Zpracování ropy doc. Ing. Josef Blažek, CSc. 6. přednáška Vlastnosti a výroba minerálních olejů ZÁKLADOVÉ OLEJE Oleje:
ODBORNÉ VZDĚLÁVÁNÍ ÚŘEDNÍKŮ PRO VÝKON STÁTNÍ SPRÁVY OCHRANY OVZDUŠÍ V ČESKÉ REPUBLICE Zpracování ropy doc. Ing. Josef Blažek, CSc. 6. přednáška Vlastnosti a výroba minerálních olejů ZÁKLADOVÉ OLEJE Oleje:
2.4 Stavové chování směsí plynů Ideální směs Ideální směs reálných plynů Stavové rovnice pro plynné směsi
 1. ZÁKLADNÍ POJMY 1.1 Systém a okolí 1.2 Vlastnosti systému 1.3 Vybrané základní veličiny 1.3.1 Množství 1.3.2 Délka 1.3.2 Délka 1.4 Vybrané odvozené veličiny 1.4.1 Objem 1.4.2 Hustota 1.4.3 Tlak 1.4.4
1. ZÁKLADNÍ POJMY 1.1 Systém a okolí 1.2 Vlastnosti systému 1.3 Vybrané základní veličiny 1.3.1 Množství 1.3.2 Délka 1.3.2 Délka 1.4 Vybrané odvozené veličiny 1.4.1 Objem 1.4.2 Hustota 1.4.3 Tlak 1.4.4
ÚPRAVA VODY V ENERGETICE. Ing. Jiří Tomčala
 ÚPRAVA VODY V ENERGETICE Ing. Jiří Tomčala Úvod Voda je v elektrárnách po palivu nejdůležitější surovinou Její množství v provozních systémech elektráren je mnohonásobně větší než množství spotřebovaného
ÚPRAVA VODY V ENERGETICE Ing. Jiří Tomčala Úvod Voda je v elektrárnách po palivu nejdůležitější surovinou Její množství v provozních systémech elektráren je mnohonásobně větší než množství spotřebovaného
EVROPSKÝ PARLAMENT C6-0267/2006. Společný postoj. Dokument ze zasedání 2003/0256(COD) 06/09/2006
 06/09/2006") EVROPSKÝ PARLAMENT 2004 Dokument ze zasedání 2009 C6-0267/2006 2003/0256(COD) CS 06/09/2006 Společný postoj Společný postoj Rady k přijetí nařízení Evropského parlamentu a Rady o registraci, hodnocení,
EVROPSKÝ PARLAMENT 2004 Dokument ze zasedání 2009 C6-0267/2006 2003/0256(COD) CS 06/09/2006 Společný postoj Společný postoj Rady k přijetí nařízení Evropského parlamentu a Rady o registraci, hodnocení,
PRŮMYSLOVÉ PROCESY. Přenos hmoty Kolony
 PRŮMYSLOVÉ PROCESY Přenos hmoty Kolony Prof. Ing. Tomáš Jirout, Ph.D. (e-mail: Tomas.Jirout@fs.cvut.cz, tel.: 2 2435 2681) DESTILACE Teoretický úvod Rovnováha neomezeně mísitelných kapalin A. Ideální chování
PRŮMYSLOVÉ PROCESY Přenos hmoty Kolony Prof. Ing. Tomáš Jirout, Ph.D. (e-mail: Tomas.Jirout@fs.cvut.cz, tel.: 2 2435 2681) DESTILACE Teoretický úvod Rovnováha neomezeně mísitelných kapalin A. Ideální chování
Směsi, roztoky. Disperzní soustavy, roztoky, koncentrace
 Směsi, roztoky Disperzní soustavy, roztoky, koncentrace 1 Směsi Směs je soustava, která obsahuje dvě nebo více chemických látek. Mezi složkami směsi nedochází k chemickým reakcím. Fyzikální vlastnosti
Směsi, roztoky Disperzní soustavy, roztoky, koncentrace 1 Směsi Směs je soustava, která obsahuje dvě nebo více chemických látek. Mezi složkami směsi nedochází k chemickým reakcím. Fyzikální vlastnosti
Teorie transportu plynů a par polymerními membránami. Doc. Ing. Milan Šípek, CSc. Ústav fyzikální chemie VŠCHT Praha
 Teorie transportu plynů a par polymerními membránami Doc. Ing. Milan Šípek, CSc. Ústav fyzikální chemie VŠCHT Praha Úvod Teorie transportu Difuze v polymerních membránách Propustnost polymerních membrán
Teorie transportu plynů a par polymerními membránami Doc. Ing. Milan Šípek, CSc. Ústav fyzikální chemie VŠCHT Praha Úvod Teorie transportu Difuze v polymerních membránách Propustnost polymerních membrán
Nauka o materiálu. Přednáška č.10 Difuze v tuhých látkách, fáze a fázové přeměny
 Nauka o materiálu Přednáška č.10 Difuze v tuhých látkách, fáze a fázové přeměny Difuze v tuhých látkách Difuzí nazýváme přesun atomů nebo iontů na vzdálenost větší než je meziatomová vzdálenost. Hnací
Nauka o materiálu Přednáška č.10 Difuze v tuhých látkách, fáze a fázové přeměny Difuze v tuhých látkách Difuzí nazýváme přesun atomů nebo iontů na vzdálenost větší než je meziatomová vzdálenost. Hnací
DESTILAČNÍ ZKOUŠKA PALIV
 VYSOKÁ ŠKOLA CHEMICKO-TECHNOLOGICKÁ V PRAZE Fakulta technologie ochrany prostředí Ústav technologie ropy a alternativních ativních paliv DESTILAČNÍ ZKOUŠKA PALIV Laboratorní cvičení ÚVOD Destilační zkouška
VYSOKÁ ŠKOLA CHEMICKO-TECHNOLOGICKÁ V PRAZE Fakulta technologie ochrany prostředí Ústav technologie ropy a alternativních ativních paliv DESTILAČNÍ ZKOUŠKA PALIV Laboratorní cvičení ÚVOD Destilační zkouška
Chromatografie. Petr Breinek
 Chromatografie Petr Breinek Chromatografie-I 2012 Společným znakem všech chromatografických metod je kontinuální dělení složek analyzované směsi mezi dvěma fázemi. Pohyblivá fáze (mobilní), eluent Nepohyblivá
Chromatografie Petr Breinek Chromatografie-I 2012 Společným znakem všech chromatografických metod je kontinuální dělení složek analyzované směsi mezi dvěma fázemi. Pohyblivá fáze (mobilní), eluent Nepohyblivá
Mol. fyz. a termodynamika
 Molekulová fyzika pracuje na základě kinetické teorie látek a statistiky Termodynamika zkoumání tepelných jevů a strojů nezajímají nás jednotlivé částice Molekulová fyzika základem jsou: Látka kteréhokoli
Molekulová fyzika pracuje na základě kinetické teorie látek a statistiky Termodynamika zkoumání tepelných jevů a strojů nezajímají nás jednotlivé částice Molekulová fyzika základem jsou: Látka kteréhokoli
Hmotnostní spektrometrie
 Hmotnostní spektrometrie Princip: 1. Ze vzorku jsou tvořeny ionty na úrovni molekul, nebo jejich zlomků (fragmentů), nebo až volných atomů dodáváním energie, např. uvolnění atomů ze vzorku nebo přímo rozštěpení
Hmotnostní spektrometrie Princip: 1. Ze vzorku jsou tvořeny ionty na úrovni molekul, nebo jejich zlomků (fragmentů), nebo až volných atomů dodáváním energie, např. uvolnění atomů ze vzorku nebo přímo rozštěpení
Vícefázové reaktory. Probublávaný reaktor plyn kapalina katalyzátor. Zuzana Tomešová
 Vícefázové reaktory Probublávaný reaktor plyn kapalina katalyzátor Zuzana Tomešová 2008 Probublávaný reaktor plyn - kapalina - katalyzátor Hydrogenace méně těkavých látek za vyššího tlaku Kolony naplněné
Vícefázové reaktory Probublávaný reaktor plyn kapalina katalyzátor Zuzana Tomešová 2008 Probublávaný reaktor plyn - kapalina - katalyzátor Hydrogenace méně těkavých látek za vyššího tlaku Kolony naplněné
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ. Stanovení těkavých látek
 LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení těkavých látek (metoda: plynová chromatografie s hmotnostně spektrometrickým detektorem) Garant úlohy: Ing. Jaromír Hradecký, Ph.D. 1 OBSAH Základní
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení těkavých látek (metoda: plynová chromatografie s hmotnostně spektrometrickým detektorem) Garant úlohy: Ing. Jaromír Hradecký, Ph.D. 1 OBSAH Základní
PROCESY V TECHNICE BUDOV 8
 UNIVERZITA TOMÁŠE BATI VE ZLÍNĚ FAKULTA APLIKOVANÉ INFORMATIKY PROCESY V TECHNICE BUDOV 8 Dagmar Janáčová, Hana Charvátová Zlín 2013 Tento studijní materiál vznikl za finanční podpory Evropského sociálního
UNIVERZITA TOMÁŠE BATI VE ZLÍNĚ FAKULTA APLIKOVANÉ INFORMATIKY PROCESY V TECHNICE BUDOV 8 Dagmar Janáčová, Hana Charvátová Zlín 2013 Tento studijní materiál vznikl za finanční podpory Evropského sociálního
www.zlinskedumy.cz Inovace výuky prostřednictvím šablon pro SŠ
 Název projektu Číslo projektu Název školy Autor Název šablony Název DUMu Stupeň a typ vzdělávání Vzdělávací oblast Vzdělávací obor Tematický okruh Inovace výuky prostřednictvím šablon pro SŠ CZ.1.07/1.5.00/34.0748
Název projektu Číslo projektu Název školy Autor Název šablony Název DUMu Stupeň a typ vzdělávání Vzdělávací oblast Vzdělávací obor Tematický okruh Inovace výuky prostřednictvím šablon pro SŠ CZ.1.07/1.5.00/34.0748
Dávkování vzorku v GC - II Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253
 Dávkování vzorku v GC - II Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 7. Dávkování ventily (Valves) Dávkovací ventily jsou jednoduchá zařízení umožňující vnesení daného objemu
Dávkování vzorku v GC - II Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 7. Dávkování ventily (Valves) Dávkovací ventily jsou jednoduchá zařízení umožňující vnesení daného objemu
Paliva. nejběžnějším zdrojem tepla musí splňovat tyto podmínky: co nejmenší náklady na těžbu a výrobu snadno uskutečnitelné spalování
 Paliva Paliva nejběžnějším zdrojem tepla musí splňovat tyto podmínky: co nejmenší náklady na těžbu a výrobu snadno uskutečnitelné spalování Dělení paliv podle skupenství pevná uhlí, dřevo kapalná benzín,
Paliva Paliva nejběžnějším zdrojem tepla musí splňovat tyto podmínky: co nejmenší náklady na těžbu a výrobu snadno uskutečnitelné spalování Dělení paliv podle skupenství pevná uhlí, dřevo kapalná benzín,
rtuť při 0 o C = 470 mn m 1 15,45 17,90 19,80 21,28
 zkapalněné plyny - velmi nízké; např. helium 0354 mn m při teplotě 270 C vodík 2 mn m při teplotě 253 C roztavené kovy - velmi vysoké; např. měď při teplotě tání = 00 mn m organické látky při teplotě 25
zkapalněné plyny - velmi nízké; např. helium 0354 mn m při teplotě 270 C vodík 2 mn m při teplotě 253 C roztavené kovy - velmi vysoké; např. měď při teplotě tání = 00 mn m organické látky při teplotě 25
ZŠ ÚnO, Bratří Čapků 1332
 Úvodní obrazovka Menu (vlevo nahoře) Návrat na hlavní stránku Obsah Výsledky Poznámky Záložky edunet Konec Chemie 1 (pro 12-16 let) LangMaster Obsah (střední část) výběr tématu - dvojklikem v seznamu témat
Úvodní obrazovka Menu (vlevo nahoře) Návrat na hlavní stránku Obsah Výsledky Poznámky Záložky edunet Konec Chemie 1 (pro 12-16 let) LangMaster Obsah (střední část) výběr tématu - dvojklikem v seznamu témat
Separační procesy Separační procesy. Dělení heterogenních směsí
 Separační procesy Separační procesy Slouží k oddělení heterogenních i homogenních směsí chemických látek na základě odlišných fyzikálně-chemických vlastností. Nejčastěji se jedná o směs produktů (hlavní
Separační procesy Separační procesy Slouží k oddělení heterogenních i homogenních směsí chemických látek na základě odlišných fyzikálně-chemických vlastností. Nejčastěji se jedná o směs produktů (hlavní
Organická chemie 1. ročník studijního oboru - gastronomie.
 Organická chemie 1. ročník studijního oboru - gastronomie. T-4 Metody oddělování složek směsí. Zpracováno v rámci projektu Zlepšení podmínek ke vzdělávání Registrační číslo projektu: CZ.1.07/1.5.00/34.0639
Organická chemie 1. ročník studijního oboru - gastronomie. T-4 Metody oddělování složek směsí. Zpracováno v rámci projektu Zlepšení podmínek ke vzdělávání Registrační číslo projektu: CZ.1.07/1.5.00/34.0639
III/2 Inovace a zkvalitnění výuky prostřednictvím ICT. Test k ověření znalostí o ropě 2. verze
 Číslo projektu CZ.1.07/1.5.00/34.0514 Číslo a název šablony klíčové aktivity III/2 Inovace a zkvalitnění výuky prostřednictvím ICT Tematická oblast Suroviny organické technologie, vy_32_inovace_ma_09_32
Číslo projektu CZ.1.07/1.5.00/34.0514 Číslo a název šablony klíčové aktivity III/2 Inovace a zkvalitnění výuky prostřednictvím ICT Tematická oblast Suroviny organické technologie, vy_32_inovace_ma_09_32
Skupenské stavy látek. Mezimolekulární síly
 Skupenské stavy látek Mezimolekulární síly 1 Interakce iont-dipól Např. hydratační (solvatační) interakce mezi Na + (iont) a molekulou vody (dipól). Jde o nejsilnější mezimolekulární (nevazebnou) interakci.
Skupenské stavy látek Mezimolekulární síly 1 Interakce iont-dipól Např. hydratační (solvatační) interakce mezi Na + (iont) a molekulou vody (dipól). Jde o nejsilnější mezimolekulární (nevazebnou) interakci.
Teorie chromatografie - II
 Teorie chromatografie - II Příprava předmětu byla podpořena projektem OPP č. CZ.2.17/3.1.00/33253 2.2 Interakce mezi molekulami Mezi elektroneutrálními molekulami působí slabé přitažlivé síly, které sdružují
Teorie chromatografie - II Příprava předmětu byla podpořena projektem OPP č. CZ.2.17/3.1.00/33253 2.2 Interakce mezi molekulami Mezi elektroneutrálními molekulami působí slabé přitažlivé síly, které sdružují
Hmotnost atomů a molekul 6 Látkové množství 11. Rozdělení směsí 16 Separační metody 20. Hustota, hmotnostní a objemový zlomek 25.
 Obsah Obecná chemie II. 1. Látkové množství Hmotnost atomů a molekul 6 Látkové množství 11 2. Směsi Rozdělení směsí 16 Separační metody 20 3. Chemické výpočty Hustota, hmotnostní a objemový zlomek 25 Koncentrace
Obsah Obecná chemie II. 1. Látkové množství Hmotnost atomů a molekul 6 Látkové množství 11 2. Směsi Rozdělení směsí 16 Separační metody 20 3. Chemické výpočty Hustota, hmotnostní a objemový zlomek 25 Koncentrace
Základní škola a mateřská škola Hutisko Solanec. žák uvede základní druhy uhlovodíků, jejich použití a zdroje. Chemie - 9. ročník
 Základní škola a mateřská škola Hutisko Solanec Digitální učební materiál Anotace: Autor: Jazyk: Očekávaný výstup: Speciální vzdělávací potřeby: Klíčová slova: Druh učebního materiálu: Druh interaktivity:
Základní škola a mateřská škola Hutisko Solanec Digitální učební materiál Anotace: Autor: Jazyk: Očekávaný výstup: Speciální vzdělávací potřeby: Klíčová slova: Druh učebního materiálu: Druh interaktivity:
Gymnázium a Střední odborná škola, Rokycany, Mládežníků 1115
 Gymnázium a Střední odborná škola, Rokycany, Mládežníků 1115 Číslo projektu: Číslo šablony: CZ.1.07/1.5.00/34.0410 III/2 Inovace a zkvalitnění výuky prostřednictvím ICT. Název materiálu: Fosilní zdroje
Gymnázium a Střední odborná škola, Rokycany, Mládežníků 1115 Číslo projektu: Číslo šablony: CZ.1.07/1.5.00/34.0410 III/2 Inovace a zkvalitnění výuky prostřednictvím ICT. Název materiálu: Fosilní zdroje
Chemie - 3. ročník. přesahy, vazby, mezipředmětové vztahy průřezová témata. očekávané výstupy RVP. témata / učivo. očekávané výstupy ŠVP.
 očekávané výstupy RVP témata / učivo Chemie - 3. ročník Žák: očekávané výstupy ŠVP přesahy, vazby, mezipředmětové vztahy průřezová témata 1.1., 1.2., 1.3., 1.4., 2.1. 1. Látky přírodní nebo syntetické
očekávané výstupy RVP témata / učivo Chemie - 3. ročník Žák: očekávané výstupy ŠVP přesahy, vazby, mezipředmětové vztahy průřezová témata 1.1., 1.2., 1.3., 1.4., 2.1. 1. Látky přírodní nebo syntetické
Průtokové metody (Kontinuální měření v proudu kapaliny)
") Průtokové metody (Kontinuální měření v proudu kapaliny) 1. Přímé měření: analyzovaná kapalina většinou odvětvena + vhodný detektor 2. Kapalinová chromatografie (HPLC) Stanovení po předchozí separaci 3.
Průtokové metody (Kontinuální měření v proudu kapaliny) 1. Přímé měření: analyzovaná kapalina většinou odvětvena + vhodný detektor 2. Kapalinová chromatografie (HPLC) Stanovení po předchozí separaci 3.
Odmašťování rozpouštědly znamená obvykle použití chlorovaných uhlovodíků (CHC dnes jen v uzavřených zařízeních), alkoholů, terpenů, ketonů, benzínu,
, alkoholů, terpenů, ketonů, benzínu,") Kubíček J. FSI 2018 Odmašťování velmi důležitá operace: odstranění tuků, prachových částic, zbytků po tryskání, kovové třísky a vody. Nečistoty jsou vázány fyzikální adsorpcí a adhezními silami. Odmašťování
Kubíček J. FSI 2018 Odmašťování velmi důležitá operace: odstranění tuků, prachových částic, zbytků po tryskání, kovové třísky a vody. Nečistoty jsou vázány fyzikální adsorpcí a adhezními silami. Odmašťování
různorodé suspenze (pevná látka v kapalné) emulze (nemísitelné kapaliny) pěna (plynná l. v kapalné l.) mlha (kapalná l. v plynné l.
 emulze (nemísitelné kapaliny) pěna (plynná l. v kapalné l.) mlha (kapalná l. v plynné l.") Obsah: 6_Směsi... 2 7_Roztoky, složení roztoku... 3 8_PL_Složení roztoku - příklady... 4 9_Rozpustnost látky... 8 10_ PL_Rozpustnost ve vodě... 9 11_ Dělení směsí... 11 1 6_ Směsi - jsou látky složené
Obsah: 6_Směsi... 2 7_Roztoky, složení roztoku... 3 8_PL_Složení roztoku - příklady... 4 9_Rozpustnost látky... 8 10_ PL_Rozpustnost ve vodě... 9 11_ Dělení směsí... 11 1 6_ Směsi - jsou látky složené
VYSOKOÚČINNÁ DESTILACE DVOUSLOŽKOVÉ SMĚSI, VÝPOČET ÚČINNOSTI
 VYSOKÁ ŠKOLA CHEMICKO-TECHNOLOGICKÁ V PRAZE Fakulta technologie ochrany prostředí Ústav technologie ropy a alternativních paliv VYSOKOÚČINNÁ DESTILACE DVOUSLOŽKOVÉ SMĚSI, VÝPOČET ÚČINNOSTI Laboratorní
VYSOKÁ ŠKOLA CHEMICKO-TECHNOLOGICKÁ V PRAZE Fakulta technologie ochrany prostředí Ústav technologie ropy a alternativních paliv VYSOKOÚČINNÁ DESTILACE DVOUSLOŽKOVÉ SMĚSI, VÝPOČET ÚČINNOSTI Laboratorní
Základy chemických technologií
 8. Přednáška Extrakce Sušení Extrakce extrakce kapalina kapalina rovnováha kapalina kapalina pro dvousložkové systémy jednostupňová extrakce, opakovaná extrakce procesní zařízení extrakce kapalina pevná
8. Přednáška Extrakce Sušení Extrakce extrakce kapalina kapalina rovnováha kapalina kapalina pro dvousložkové systémy jednostupňová extrakce, opakovaná extrakce procesní zařízení extrakce kapalina pevná
Ropa Kondenzované uhlovodíky
 Nejdůležitější surovina pro výrobu organických sloučenin Nejvýznamnější surovina světové ekonomiky Výroba energie Chemické zpracování - 15 % Cena a zásoby ropy (70-100 let) Ropné krize Nutnost hledání
Nejdůležitější surovina pro výrobu organických sloučenin Nejvýznamnější surovina světové ekonomiky Výroba energie Chemické zpracování - 15 % Cena a zásoby ropy (70-100 let) Ropné krize Nutnost hledání
Simulovaná destilace ropných frakcí
 Středisko analytické chemie pracoviště Litvínov Návod na laboratorní práci Simulovaná destilace ropných frakcí Simulovaná destilace středních destilátů a vakuových destilátů pomocí plynové chromatografie,
Středisko analytické chemie pracoviště Litvínov Návod na laboratorní práci Simulovaná destilace ropných frakcí Simulovaná destilace středních destilátů a vakuových destilátů pomocí plynové chromatografie,
Metody separace. přírodních látek
 Metody separace přírodních látek (5) Chromatografie; základní definice a klasifikace ruzných metod; kapalinová chromatografie, plynová chromatografie, přístrojová technika. Chromatografie «F(+)d» 1897
Metody separace přírodních látek (5) Chromatografie; základní definice a klasifikace ruzných metod; kapalinová chromatografie, plynová chromatografie, přístrojová technika. Chromatografie «F(+)d» 1897
ZŠ ÚnO, Bratří Čapků 1332
 Animovaná chemie Top-Hit Analytická chemie Analýza anorganických látek Důkaz aniontů Důkaz kationtů Důkaz kyslíku Důkaz vody Gravimetrická analýza Hmotnostní spektroskopie Chemická analýza Nukleární magnetická
Animovaná chemie Top-Hit Analytická chemie Analýza anorganických látek Důkaz aniontů Důkaz kationtů Důkaz kyslíku Důkaz vody Gravimetrická analýza Hmotnostní spektroskopie Chemická analýza Nukleární magnetická
Krása fázových diagramů jak je sestrojit a číst Silvie Mašková
 Krása fázových diagramů jak je sestrojit a číst Silvie Mašková Katedra fyziky kondenzovaných látek Matematicko-fyzikální fakulta Univerzita Karlova Praha Pár základích pojmů na začátek Co jsou fázové diagramy?
Krása fázových diagramů jak je sestrojit a číst Silvie Mašková Katedra fyziky kondenzovaných látek Matematicko-fyzikální fakulta Univerzita Karlova Praha Pár základích pojmů na začátek Co jsou fázové diagramy?
Při reálném chromatografickém ději nikdy nedojde k ustavení rovnováhy mezi oběma fázemi První ucelená teorie respektující uvedenou skutečnost byla
 Teorie chromatografie - III Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 4.3.3 Teorie dynamická Při reálném chromatografickém ději nikdy nedojde k ustavení rovnováhy mezi oběma
Teorie chromatografie - III Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 4.3.3 Teorie dynamická Při reálném chromatografickém ději nikdy nedojde k ustavení rovnováhy mezi oběma
Třífázové trubkové reaktory se zkrápěným ložem katalyzátoru. Předmět: Vícefázové reaktory Jméno: Veronika Sedláková
 Třífázové trubkové reaktory se zkrápěným ložem katalyzátoru Předmět: Vícefázové reaktory Jméno: Veronika Sedláková 3-fázové reakce Autoklávy (diskontinuální) Trubkové reaktory (kontinuální) Probublávané
Třífázové trubkové reaktory se zkrápěným ložem katalyzátoru Předmět: Vícefázové reaktory Jméno: Veronika Sedláková 3-fázové reakce Autoklávy (diskontinuální) Trubkové reaktory (kontinuální) Probublávané
Fyzikální chemie. Magda Škvorová KFCH CN463 magda.skvorova@ujep.cz, tel. 3302. 14. února 2013
 Fyzikální chemie Magda Škvorová KFCH CN463 magda.skvorova@ujep.cz, tel. 3302 14. února 2013 Co je fyzikální chemie? Co je fyzikální chemie? makroskopický přístup: (klasická) termodynamika nerovnovážná
Fyzikální chemie Magda Škvorová KFCH CN463 magda.skvorova@ujep.cz, tel. 3302 14. února 2013 Co je fyzikální chemie? Co je fyzikální chemie? makroskopický přístup: (klasická) termodynamika nerovnovážná
Stanovení složení mastných kyselin
 LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení složení mastných kyselin (metoda: plynová chromatografie s plamenovým ionizačním detektorem) Garant úlohy: Ing. Jana Kohoutková, Ph.D. 1 Obsah
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení složení mastných kyselin (metoda: plynová chromatografie s plamenovým ionizačním detektorem) Garant úlohy: Ing. Jana Kohoutková, Ph.D. 1 Obsah
Fyzikální chemie. ochrana životního prostředí analytická chemie chemická technologie denní. Platnost: od 1. 9. 2009 do 31. 8. 2013
 Učební osnova předmětu Fyzikální chemie Studijní obor: Aplikovaná chemie Zaměření: Forma vzdělávání: Celkový počet vyučovacích hodin za studium: Analytická chemie Chemická technologie Ochrana životního
Učební osnova předmětu Fyzikální chemie Studijní obor: Aplikovaná chemie Zaměření: Forma vzdělávání: Celkový počet vyučovacích hodin za studium: Analytická chemie Chemická technologie Ochrana životního
Třídění látek. Chemie 1.KŠPA
 Třídění látek Chemie 1.KŠPA Systém (soustava) Vymezím si kus prostoru, látky v něm obsažené nazýváme systém soustava okolí svět Stěny soustavy Soustava může být: Izolovaná = stěny nedovolí výměnu částic
Třídění látek Chemie 1.KŠPA Systém (soustava) Vymezím si kus prostoru, látky v něm obsažené nazýváme systém soustava okolí svět Stěny soustavy Soustava může být: Izolovaná = stěny nedovolí výměnu částic
Omezování plynných emisí. Ochrana ovzduší ZS 2012/2013
 Omezování plynných emisí Ochrana ovzduší ZS 2012/2013 1 Úvod Různé fyzikální a chemické principy + biotechnologie Principy: absorpce adsorpce oxidace a redukce katalytická oxidace a redukce kondenzační
Omezování plynných emisí Ochrana ovzduší ZS 2012/2013 1 Úvod Různé fyzikální a chemické principy + biotechnologie Principy: absorpce adsorpce oxidace a redukce katalytická oxidace a redukce kondenzační
PROCESNÍ OLEJE PRO VÝROBCE PNEUMATIK
 PROCESNÍ OLEJE PRO VÝROBCE PNEUMATIK POPIS PRODUKTU MES 15 Procesní olej s označením MES 15 je olej určený jako změkčovadlo pro výrobu SBR kaučuků a jejich směsí používaných pro výrobu moderních pneumatik.
PROCESNÍ OLEJE PRO VÝROBCE PNEUMATIK POPIS PRODUKTU MES 15 Procesní olej s označením MES 15 je olej určený jako změkčovadlo pro výrobu SBR kaučuků a jejich směsí používaných pro výrobu moderních pneumatik.
Gymnázium Jiřího Ortena, Kutná Hora
 Předmět: Náplň: Třída: Počet hodin: Pomůcky: Chemie (CHE) Obecná chemie, anorganická chemie Tercie 2 hodiny týdně Školní tabule, interaktivní tabule, Apple TV, tablety, tyčinkové a kalotové modely molekul,
Předmět: Náplň: Třída: Počet hodin: Pomůcky: Chemie (CHE) Obecná chemie, anorganická chemie Tercie 2 hodiny týdně Školní tabule, interaktivní tabule, Apple TV, tablety, tyčinkové a kalotové modely molekul,
Příprava materiálu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253
 Příprava materiálu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Část 16 Iontová chromatografie Iontová chromatografie je speciální technika vyvinutá pro separaci anorganických iontů a organických
Příprava materiálu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Část 16 Iontová chromatografie Iontová chromatografie je speciální technika vyvinutá pro separaci anorganických iontů a organických
Přírodní zdroje uhlovodíků
 Tento výukový materiál vznikl za přispění Evropské unie, státního rozpočtu ČR a Středočeského kraje Říjen 2010 Mgr. Alena Jirčáková Zemní plyn - vznik: Výskyt často spolu s ropou (naftový zemní plyn) nebo
Tento výukový materiál vznikl za přispění Evropské unie, státního rozpočtu ČR a Středočeského kraje Říjen 2010 Mgr. Alena Jirčáková Zemní plyn - vznik: Výskyt často spolu s ropou (naftový zemní plyn) nebo
Očekávané výstupy podle RVP ZV Učivo předmětu Přesahy a vazby
 Předmět: CHEMIE Ročník: 8. Časová dotace: 2 hodiny týdně Očekávané výstupy podle RVP ZV Učivo předmětu Přesahy a vazby Konkretizované tematické okruhy realizovaného průřezového tématu září orientuje se
Předmět: CHEMIE Ročník: 8. Časová dotace: 2 hodiny týdně Očekávané výstupy podle RVP ZV Učivo předmětu Přesahy a vazby Konkretizované tematické okruhy realizovaného průřezového tématu září orientuje se
Do známky zkoušky rovnocenným podílem započítávají získané body ze zápočtového testu.
 Podmínky pro získání zápočtu a zkoušky z předmětu Chemicko-inženýrská termodynamika pro zpracování ropy Zápočet je udělen, pokud student splní zápočtový test alespoň na 50 %. Zápočtový test obsahuje 3
Podmínky pro získání zápočtu a zkoušky z předmětu Chemicko-inženýrská termodynamika pro zpracování ropy Zápočet je udělen, pokud student splní zápočtový test alespoň na 50 %. Zápočtový test obsahuje 3
EU peníze středním školám digitální učební materiál
 EU peníze středním školám digitální učební materiál Číslo projektu: Číslo a název šablony klíčové aktivity: Tematická oblast, název DUMu: Autor: CZ.1.07/1.5.00/34.0515 III/2 Inovace a zkvalitnění výuky
EU peníze středním školám digitální učební materiál Číslo projektu: Číslo a název šablony klíčové aktivity: Tematická oblast, název DUMu: Autor: CZ.1.07/1.5.00/34.0515 III/2 Inovace a zkvalitnění výuky
Klinická a farmaceutická analýza. Petr Kozlík Katedra analytické chemie
 Klinická a farmaceutická analýza Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz http://web.natur.cuni.cz/~kozlik/ 1 Spojení separačních technik s hmotnostní spektrometrem Separační
Klinická a farmaceutická analýza Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz http://web.natur.cuni.cz/~kozlik/ 1 Spojení separačních technik s hmotnostní spektrometrem Separační
Ultrastopová laboratoř České geologické služby
 Ultrastopová laboratoř České geologické služby Jitka Míková Česká geologická služba Praha - Barrandov Laboratorní koloběh Zadavatel TIMS Analýza vzorku Vojtěch Erban Jakub Trubač Lukáš Ackerman Jitka Míková
Ultrastopová laboratoř České geologické služby Jitka Míková Česká geologická služba Praha - Barrandov Laboratorní koloběh Zadavatel TIMS Analýza vzorku Vojtěch Erban Jakub Trubač Lukáš Ackerman Jitka Míková
VÝUKOVÝ MODUL MEMBRÁNOVÝCH PROCESŮ TÉMATA PŘEDNÁŠEK
 VÝUKOVÝ MODUL MEMBRÁNOVÝCH PROCESŮ TÉMATA PŘEDNÁŠEK TRANSPORT LÁTEK MEMBRÁNAMI Transport látek porézními membránami - Plouživý tok nestlačitelných tekutin vrstvou částic - Plouživý tok stlačitelných tekutin
VÝUKOVÝ MODUL MEMBRÁNOVÝCH PROCESŮ TÉMATA PŘEDNÁŠEK TRANSPORT LÁTEK MEMBRÁNAMI Transport látek porézními membránami - Plouživý tok nestlačitelných tekutin vrstvou částic - Plouživý tok stlačitelných tekutin
Bilan a ce c zák á l k ad a ní pojm j y m aplikace zákonů o zachování čehokoli 10.10.2008 3
 Výpočtový seminář z Procesního inženýrství podzim 2008 Bilance Materiálové a látkové 10.10.2008 1 Tématické okruhy bilance - základní pojmy bilanční schéma způsoby vyjadřování koncentrací a přepočtové
Výpočtový seminář z Procesního inženýrství podzim 2008 Bilance Materiálové a látkové 10.10.2008 1 Tématické okruhy bilance - základní pojmy bilanční schéma způsoby vyjadřování koncentrací a přepočtové
www.zlinskedumy.cz Inovace výuky prostřednictvím šablon pro SŠ
 Název projektu Číslo projektu Název školy Autor Název šablony Název DUMu Inovace výuky prostřednictvím šablon pro SŠ CZ.1.07/1.5.00/34.0748 Gymnázium Jana Pivečky a Střední odborná škola Slavičín Mgr.
Název projektu Číslo projektu Název školy Autor Název šablony Název DUMu Inovace výuky prostřednictvím šablon pro SŠ CZ.1.07/1.5.00/34.0748 Gymnázium Jana Pivečky a Střední odborná škola Slavičín Mgr.