P ř í r o d o v ě d e c k á f a k u l t a Katedra buněčné biologie a genetiky
|
|
|
- Milan Dostál
- před 5 lety
- Počet zobrazení:
Transkript
1 U N I V E R Z I T A P A L A C K É H O V O L O M O U C I P ř í r o d o v ě d e c k á f a k u l t a Katedra buněčné biologie a genetiky Molekulární patogeneze vybraných vrozených poruch produkce hemoglobinu Rigorózní práce Mgr. et Mgr. Martina Partschová Olomouc 2017
2 Prohlašuji, ţe jsem rigorózní práci zpracovala samostatně a veškerá pouţitá literatura je v ní ádně uvedena. Ve Svésedlicích dne
3 PODĚKOVÁNδ Na tomto místě bych chtěla poděkovat Doc. RNDr. Vladimíru Divokému, Ph.D a Mgr. Renátě εojzíkové, Ph.D za kvalitní odborné vedení, mnoho cenných rad a p ipomínek p i ešení mé práce. Děkuji všem koleg m z Ústavu biologie LF UP za vytvo ení dobrého pracovního prost edí a pomoc p i ešení odborných problém. Zvláště bych chtěla poděkovat vedoucí laborato e molekulární biologie na HOK FN Olomouc RNDr. εartině Divoké, Ph.D. za poskytnutí vzork DNů od pacient, za její cenné rady a konzultace p i ešení této práce. Děkuji také své rodině za jejich nesmírnou trpělivost, toleranci a všestrannou podporu.
4 SOUHRN Poruchy produkce hemoglobinu jsou nejčastějšími poruchami krvetvorby. Poruchy produkce hemoglobinu mohou být vrozené nebo získané; vrozenými p íčinami jsou mutace narušující syntézu globin nebo metabolizmus hemoglobinu a mutace gen zapojených do p íjmu iont ţeleza. V této práci jsme se soust edili na odhalování molekulární talasemie, nejčastější vrozené hypochromní mikrocytární anémie v české a slovenské populaci. Během 8 let jsme diagnostikovali 1Řγ pacient ze 10δ nep íbuzných rodin s talasemií. Na molekulární úrovni jsme identifikovali 16 r zných -talasemických alel. Všichni -talasemičtí pacienti byli heterozygoti pro mutaci v HBB genu, manifestující talasemii minor. Tato práce také p ináší srovnání s d íve publikovanými studiemi na stejné téma. Dále jsme u t ech pacient diagnostikovali enzymový typ methemoglobinémie prokázáním heterozygotní mutace v genu CYB5R3 mající za následek sníţení aktivity enzymu cytochrom-b5-reduktázy, který se podílí na redukci methemoglobinu v erytrocytech. V rámci skupiny pacient s deficitem pyruvátkinázy jsme u neonatální hyperferitinémie asociované s novou posunovou delecí v genu PKLR vyloučili kauzální mutaci pro primární hemochromatózu.
5 SUMMARY Disorders of hemoglobin production are the most common defects of hematopoiesis. Disorders of hemoglobin production are hereditary or acquired; among inherited genetic defects are the most common mutations of globin synthesis, mutations affecting hemoglobin metabolism and mutations of genes involved in iron ion acquisition and utilization. In this study, we were focused on molecular etiology of -thalassemia, the main cause of a congenital hypochromic microcytic anemia in the Czech and Sovak populations. During the last 8 years we have diagnosed 1Řγ patients from 10δ unrelated families with thalassemia. At the molecular level we identified 16 different -thalassemia alleles. ůll thalassemia patients were heterozygous for mutation in the HBB gene, manifesting thalassemia minor. This work also provides a comparison with previously published studies on the same topic. Further, we diagnosed three patients to have enzyme type of methemoglobinemia due to heterozygous mutations in the CYB5R3 gene, which affect activity of cytochromeb5-reductase responsible for methemoglobin reduction in red blood cells. Among pyruvate kinase deficient patients we decribed the first case with neonatal hyperferritinemia associated with a novel frameshift deletion in the PKLR gene and excluded genetic causes for primary hemochromatosis in this patient.
6
7 OBSAH ÚVOD... 8 TEORETICKÁ ČÁST Hematopoéza Erytropoéza Hemoglobin Syntéza hemoglobinu Druhy hemoglobinu Hemoglobinopatie Strukturální hemoglobinové varianty Hereditární perzistence fetálního hemoglobinu Talasemie α talasemie talasemie Klinická klasifikace talasemií Patofyziologie talasemií Diagnostika Výskyt talasemií talasemie a talasemie εethemoglobinémie Deficit cytochrom-b5-reduktázy Hemoglobin M P etíţení organismu ţelezem Ţelezo v lidském těle εetabolismus ţeleza Regulace homeostázy ţeleza Primární hemochromatóza Sekundární hemochromatóza Cíl práce EXPERIεENTÁδNÍ ČÁST εateriál
8 7.1. Biologický materiál Chemikálie a roztoky P ístrojové vybavení laborato e a software Pracovní postupy εolekulárně genetická analýza Izolace DNA z krve (fenol-chloroformová extrakce) PCR Sekvenování DNů Enzymová analýza P íprava lyzátu pro enzymovou analýzu Spektrofotometrické stanovení aktivity cytochrom-b5-reduktázy Stanovení koncentrace hemoglobinu v enzymovém lyzátu Výsledky talasemie εethemoglobinémie Hemochromatóza Diskuze Závěr SEZNAM OBRÁZK SEZNAM TABULEK SEZNůε POUŢITÝCH ZKRůTEK SEZNAM LITERATURY SEZNůε P ÍδOH P ÍδOHY 7
9 ÚVOD Produkce hemoglobinu v erytroidních buňkách je závislá na t ech r zných procesech: syntéze globin, syntéze protoporfyrinu (hemového prekurzoru) a dalších metabolických drahách hemoglobinu a na p íjmu a utilizaci iont ţeleza. Všechny t i procesy musí být p ísně regulovány a koordinovány, protoţe porucha této regulace m ţe mít za následek vznik patologických stav jako jsou anémie, porfýrie, hemachromatóza či naopak polycytémie. ada těchto patologických stav je zp sobena vrozenými mutacemi gen účastnících se globinové nebo hemové syntézy či metabolizmu iont ţeleza. Z těchto vrozených mutací jsou nejčastější ty, které zp sobují vrozené poruchy syntézy nebo struktury samotného hemoglobinu, hemoglobinopatie. Talasemie, které se adí mezi hemoglobinopatie, jsou povaţovány za nejčastější geneticky monogenně podmíněné choroby na světě autosomálně recesivního charakteru (Weatherall, 2006). Vznikají v d sledku poruchy syntézy α nebo (vzácněji nebo ) globinového etězce a pat í tedy ke kvantitativním poruchám nikoli kvalitativním jako je tomu v p ípadě strukturních hemoglobinových variant. Je známo více neţ β00 mutací globinového genu ( které mohou vést k talasemiím a globinovým strukturním variantám. Některé mutace mohou zp sobit poruchu s charakteristikami jak talasemie tak globinové varianty. Talasemie je rozší ena zejména v tzv. malarických oblastech, tj. v oblastech kolem St edozemního mo e, na Blízkém východě, v Indii a jihovýchodní ůsii. P estoţe jsou a -talasemické alely ve st ední Evropě relativně vzácné, jsou v České i Slovenské republice nejčastější p íčinou vrozené mikrocytární anémie (Divoký et al., 2005). Tato skutečnost je p ipisována jednak historickým vliv m (kontakty s národy St edomo í v Rakouském císa ství a v Rakousku-Uhersku), tak p edevším nar stající migraci obyvatelstva. V České republice pat í mezi respektovaná centra s dlouholetou zkušeností ve studiu vrozených poruch erytropoézy Hemato-onkologická klinika, Dětská klinika a Ústav biologie LF UP a FN v Olomouci. Problematice talasemií se zde začal věnovat nejprve prof. K. Indrák a následně i prof. D. Pospíšilová a doc. V. Divoký, kte í dovedli diagnostiku talasemií na světovou úroveň. 8
10 Vrozená methemoglobinémie typu I a II je autosomálně recesivním onemocněním, které je zp sobené deficitem enzymu cytochrom-b5-reduktázy podílejícího se na zpětné redukci methemoglobinu. Týká-li se deficit pouze erytrocyt, pak hovo íme o typu I; týkáli se všech buněk, pak se jedná o typ II. Rozlišujeme rovněţ vrozenou methemoglobinémii typu III, která je však autosomálně dominantního charakteru a vzniká v d sledku p ítomnosti hemoglobinové varianty tzv. hemoglobinu M. V České republice se spíše setkáváme s methemoglobinémiemi asociovanými s jinými patologickými stavy (vrozeného charakteru) nebo získanými formami. V p ípadě neefektivní erytropoézy nebo anémie se často setkáváme s p etíţením organizmu ţelezem. Nemocní s hereditární hemochromatózou nejsou schopni zvýšit expresi hepcidinu v odpovědi na p etíţení ţelezem, nejsou tudíţ schopni degradovat feroportin a zastavit další p ísun ţeleza z potravy do organismu. Ke vzniku tzv. sekundární hemochromatózy pak p ispívají další faktory jako je transfuze, splenektomie nebo koincidence s primární vrozenou hemochromatózou. Ta se dělí na δ podtypy I aţ IV, podle toho, zda se kauzální mutace nachází v genu HFE (nejčastější varianta, kódující protein HFE), HFE2 (hemojuvelin) nebo HAMP (hepcidin), TFR2 (transferinový receptor β) či SLC40A1 (feroportin). P edkládaná rigorózní práce je zamě ena na problematiku molekulární patogeneze vybraných vrozených poruch hemoglobinové syntézy. Největší díl mé práce je věnován talasemiím, v menší mí e se soust edí na problematiku methemoglobinémie a hemochromatóz. 9
11 TEORETICKÁ ČÁST 1. Hematopoéza Hematopoéza (krvetvorba) je nesmírně komplikovaný, komplexně ízený a dodnes ne zcela prozkoumaný proces (Ketley et Newland, 1997). P edstavuje tvorbu krevních buněk v krvetvorných orgánech. Probíhá po celý ţivot člověka. Z hlediska vývoje lidského organismu rozeznáváme t i stádia krvetvorby: 1) mezoblastové, β) hepatolineální, a γ) medulární. Mezoblastové období krvetvorby začíná mezi 1δ. a 1ř. dnem nitroděloţního ţivota v tzv. krevních ostr vcích ţloutkového vaku. Krvetvorba je v tomto období tak ka výhradně erytroidní a vyznačuje se tvorbou velkých normoblast a vyplavováním červených krvinek obsahujících jádro, na základě čeho je tento proces označován jako primitivní hematopoéza. Hepatolineální období nastupuje mezi prvním a druhým měsícem těhotenství, kdy se rozvíjí krvetvorba v játrech a z menší části také ve slezině. Krvetvorba ve fetálních játrech ustává v období těsně po narození. Je rovněţ podstatou erytroidní (erytrocyty jsou jiţ bezjaderné), má však jiţ normoblastický charakter. Od γ. měsíce nastupuje i granulopoéza a tvorba megakaryocyt. Období medulární (d eňové) začíná od 10. týdne intrauterinního vývoje a to nejprve v klíčních kostech, pak ve všech kostech a později pouze v plochých kostech (Krahulcová et al., 1řř6). ůţ na lymfatické orgány a lymfatickou tkáň, kde nadále probíhá lymfopoéza, nahradí d eňová krvetvorba krvetvornou činnost jiných orgán. Za některých patologických stav (hemolýza, myeloproliferativní onemocnění aj.) dochází k obnovení mimod eňové krvetvorby v játrech, slezině a dokonce i v mízních uzlinách. D eňová krvetvorba p edstavuje proces, během kterého pluripotentní hematopoetická kmenová buňka (HSC, hematopoietic stem cell) pro všechny nelymfoidní i lymfoidní ady postupně diferencuje do pluri-, bi-, monopotentních progenitorových a prekurzorových kmenových buněk, které jsou výchozími buňkami pro p íslušnou adu (Obr. 1) (Muirhead et al., 1995). εultipotentní kmenová buňka pro všechny nelymfoidní ady CFU-GEMM 10
12 (colony forming unit granulocytes, erythrocytes, monocytes/macrophages, megakaryocytes) se postupně diferencuje do granulocytové, erytroidní, monocytové a megakaryocytové ady. Erytropoéza a megakaryopoéza vychází ze společné bipotentní kmenové buňky εep (megakaryocyte/erythroid progenitor). Erytropoéza se pak diferencuje od BFU-E (burst forming unit-erythroid) p es CFU-E k proerytroblast m. Megakaryocytární linie se vyvíjí z kmenové buňky CFU-εeg, která se pak diferencuje v megakaryoblasty, promegakaryocyty a megakaryocyty, z kterých vznikají trombocyty. Granulopoéza a monocytopoéza vychází ze společné kmenové buňky CFU-GM. δymfoidní ada vychází ze společné kmenové buňky pro B- a T- adu a NK-buňky ze specifického NK-prekurzoru (Melchers et Rolink, 2001; Sharpless et DePinho, 2007). Obr. 1: Zjednodušené schéma hematopoézy (p evzato z Z hematopoetické kmenové buňky (HSC hemopoietic stem cell) vznikají multipotentní progenitory a následně oligopotentní progenitory, mezi které pat í společný lymfoidní progenitor (CδP common lymphoid progenitor) a společný myeloidní progenitor (CεP common myeloid progenitor). Z CεP se vyvíjí p es multipotentní kmenovou buňku CFU-GEMM (colony forming unit granulocytes, erythrocytes, monocytes/macrophages, megakaryocytes) dále progenitor granulocyt a makrofág (GεP granulocyte-macrophage progenitor) a progenitor megakaryocyt a erytrocyt (εep megakaryocyte-erythrocyte progenitor), které dále poskytují prekurzory jednotlivých krevních ad. V p ípadě CδP jsou to B a T lymfocyty a NK buňky, GεP tvo í monocyty, makrofágy a granulocyty, MEP poskytuje erytrocyty a megakaryocyty, které produkují krevní destičky (Melchers et Rolink, 2001; Sharpless et DePinho, 2007). 11
13 Krvetvorba je pod kontrolou ady interleukin a CFS (colony-stimulating factors). Z interleukin je to nap. interleukin γ (stimuluje progenitorové buňky všech ad), interleukin δ (r stový faktor pro B-lymfocyty), interleukin 5 (stimuluje diferenciaci B lymfocyt a tvorbu specifických protilátek), z faktor pak zejména erytropoietin vedoucí k diferenciaci do červené ady, Gε-CSF (granulo-markofágové kolonie stimulující faktor) nebo G-CSF (granulocytové kolonie stimulující faktor) Erytropoéza Erytropoéza je proces tvorby a vývoje červené krvinky (erytrocytu) v červené (erytroidní) vývojové krevní adě. Erytropoéza p edstavuje první diferencovanou hematopoetickou linii, která se objevuje během ontogeneze v krevních ostr vcích ţloutkového váčku. Na ízení erytropoézy se podílí cirkulující hormon glykoproteinové povahy erytropoetin (EPO) produkovaný v dospělosti hlavně v ledvinách, který podněcuje diferenciaci BFU-E a CFU-E v erytrocyty. V erytropoéze zastávají významnou úlohu některé vitamíny (C, B6, B1β), kyselina listová, kovy (Fe, Co, Cu) a další látky a stopové prvky. V rámci erytropoézy rozlišujeme několik erytrocytárních stádií. Z proerytroblastu (15 µm) se vyvíjí další maturací bazofilní, polychromatofilní a ortochromní erytroblasty, z nich pak bezjaderné retikulocyty (proerytrocyty), které jsou vyplavovány z kostní d eně do periferní krve, kde z nich vznikají zralé erytrocyty (7 µm). Bikonkávní tvar erytrocytu je podmíněn strukturou a vlastnostmi jejich membrány, jejichţ další funkcí je transport plyn a iont. Energetická rovnováha je v erytrocytu udrţována zejména prost ednictvím Embden-Mayerhoffovy dráhy (glykolýza) a hexozomonofosfátového cyklu. Glykolýzou vzniká kromě ůtp také β,γ-bisfosfogycerát (β,γ-bpg, spolu s Bohrovým efektem sniţuje afinitu hemoglobinu ke kyslíku v tkáních) či NůDH (redukce methemoglobinu). V p ípadě membránových (nap. sférocytózy) nebo enzymových abnormalit (enzymopatie deficit glukóza-6-fosfátdehydrogenázy, pyruvátkinázy) dochází k p edčasnému rozkladu erytrocytu hemolýze. Vývoj erytroblast trvá p ibliţně 7 dní, doba ţivota erytrocytu v periférní krvi p ibliţně 1β0 dní neţ je z d vodu ztráty plasticity fagocytována makrofágem ve slezině. 12
14 Zvýšený počet retikulocyt ( 15 ) se nachází fyziologicky u embrya a u novorozenc, patologicky pak nap. u hemolytické a megaloblastové anémie, p i krvácení. Sníţený počet erytrocyt ukazuje na útlum krvetvorby. Inefektivní erytropoéza je často výsledkem abnormalit (dysplasie), ke kterým dochází během vývoje erytroblast (abnormality jádra aj.) a vyskytující se zejména u myelodysplastického syndromu, ale provází i adu vrozených poruch, které se manifestují hemolytickou anémií. Mezi morfologické velikostní abnormality zralých krvinek se adí mikrocyty nebo makrocyty (mean corpuscular volume/hodnota st edního objemu erytrocytu, εcv, je pod Ř0 resp. nad ř5 fl), mezi tvarové abnormality pat í sférocyty, eliptocyty, poikilocyty aj., a mezi strukturní abnormality pak nap. erytrocyty s bazofilním tečkováním, Heinzovými tělísky, Cabotovými prstenci. Jednotlivé abnormality jsou charakteristické pro určitý typ anémie sníţená produkce erytrocyt a s tím související koncentrace hemoglobinu. Normální hodnota počtu erytrocytu v periferní krvi činí u muţ δ,γ-5,ř a u ţen γ,ř-5,2x10 12 /l (Pecka, 2002), závisí však nejen na pohlaví, ale i rase u dětí se navíc liší v jednotlivých věkových skupinách Hemoglobin δidský hemoglobin je metaloprotein o molekulové hmotnosti 6δ 500 Da, který je tvo en δ globinovými etězci (bílkovinná část) a prostetickou (nebílkovinnou) skupinou hemem (protoporfyrin) (Obr. 2). Tvo í aţ ř6 % pevné sloţky erytrocytu - jeden erytrocyt tak obsahuje p ibliţně γx10 8 molekul hemoglobinu tj pg (MCH, mean corpuscular hemoglobin/st ední hmotnost hemoglobinu v erytrocytu). Jeho základní funkcí je p enos plyn transport kyslíku z plic do tkání a podílí se také na p enosu oxidu uhličitého z tkání zpět do plic, p ípadně transportu oxidu dusnatého p i zánětlivých procesech (1 ml Hb je schopen vázat 1,γδ ml kyslíku). 13
. P evzato z encyklopedie Britannica, www.britannica.com. 1.3.")
, které jsou vzájemně v souladu. Zvyšováním hladiny ţeleza dochází ke stimulaci syntézy hemu a ten zase stimuluje syntézu globin.")
15 A B Obr. 2: Struktura hemoglobin a hemu. Hemoglobin je tetramer sloţený ze β r zných pár globin, které váţou 4 molekuly hemu (A). Hem je tvo ený protoporfyrinem se zabudovaným dvojmocným šestivazným ţelezem ve st edu tetrapyrolového jádra (B). P evzato z encyklopedie Britannica, Syntéza hemoglobinu Na syntéze hemoglobinu, která probíhá v nezralých formách erytrocytu, se podílejí t i buněčné procesy: syntéza globin, syntéza protoporfyrinu a p íjem iont ţeleza (dvěma posledními vzniká hem), které jsou vzájemně v souladu. Zvyšováním hladiny ţeleza dochází ke stimulaci syntézy hemu a ten zase stimuluje syntézu globin. Syntéza hemu probíhá ve všech savčích buňkách obsahujících mitochondrie, zvlášť intenzivně v erytroblastech. Základem pro syntézu protoporfyrinu je sukcinylkoenzym A a aminokyselina glycin, z kterých vzniká kyselina delta-aminolevulová (ůδů) p sobením enzymu ALA-syntetázy. Kondenzací β molekul ůδů vzniká porfobilinogen a kondenzací jeho 4 molekul pak lineární tetrapyrol cyklizující spontánně v uroporfyrinogen, který je pak p eměněn na koproporfyrinogen III, protoporfyrinogen III a metabolizován aţ na protoporfyrin. P sobením enzymu ferochelatázy (hemsyntetázy) se do protoporfyrinu včlení šestivazné ţelezo vzniká hem (Diskerson et Geis, 1983) jeden gram hemoglobinu obsahuje γ,γδ ng ţeleza. Ţelezo je čty mi vazbami vázané v tetrapyrolovém kruhu, jednou na globinový etězec a zbylá vazba je určena pro reverzibilní vazbu s kyslíkem. Začátek a konec biosyntézy hemu je lokalizován v mitochondriích, část od vzniku ůδů aţ po 14
16 koproporfyrinogen probíhá v cytosolu. Hem, který se uvolní z mitochondrie je v červené krvince inkorporován do hemoglobinu (Dickerson & Geis,1983). Syntéza globinů probíhá v endoplazmatickém retikulu zrajících erytrocyt podle obecných princip proteosyntézy. R zné peptidové etězce se syntetizují stejnou rychlostí, ačkoliv jsou geny lokalizovány na r zných chromozomech. Genová rodina (cluster) ídící syntézu α globinových etězc je lokalizována na chromozomu 16 a globinová rodina na krátkém raménku chromozomu 11 (Diesseroth et al., 1978). Cluster α globinových gen leţí blízko telomery. δokus má délku βř kb a obsahuje embryonální gen (zeta) a dva α globinové geny fetálního a dospělého typu (αβ a α1), které jsou uspo ádány na chromozomu ve směru jejich exprese během ontogeneze (Obr. 3). Kromě nich obsahuje rovněţ δ pseudogeny (zeta - pseudozeta - mí - pseudoalpha-1). Oba geny α1 a αβ se liší jen v druhém intronu (IVS-II) a ve 3 postranních oblastech. Produkované etězce jsou zcela identické αβ gen produkuje β-γkrát více α etězc neţ α1 gen. Odlišná je exprese těchto gen, kterou ídí erytroidně specifická regulační oblast nazývaná pozitivní regulační oblast HS-δ0, p sobí jako zesilovač globinové exprese (Barbour et al., 2000). Nachází se δ0 kb proti směru exprese globinového genu. globinový cluster má délku 70 kb a obsahuje 5 funkčních gen (, G, A, a ), které jsou uspo ádány ve směru jejich postupného zapínání během ontogeneze (Obr. 3). Zdraví dospělí jedinci mají (epsilon) a (gamma) globinové geny vypnuté. Geny globinové rodiny se nacházejí v oblasti chromatinu, která má charakter uzav ené domény v neerytroidní tkáni a je p ístupná transkripčním faktor m jen v erytroidních buňkách. 15
17 A) B) Obr. 3: A) Schematické znázornění α globinového lokusu na chromozomu 16 a globinového lokusu na chromozomu 11 (Nussbaum et al., 2004). B) Exprese globinového genu (upraveno dle Hoffbrand et Pettit, 1988). εolekula globinu se syntetizuje RNů transkripcí. Následně dochází k úpravám vznikající prernů. Sest ihem se odstraní p ebytečná RNů vzniklá p epsáním intron. Spojení exon/intron mají na 5 konci sekvenci GT a na konci γ sekvenci ůg. Tyto sekvence jsou d leţité pro správný sest ih. mrnů je dále modifikována navázáním 7-methylguanozinu tzv. 5 čepičky (cap) na 5 konci, která je d leţitá pro rozpoznání mrnů ribozomem, a p idáním poly-ů konce (β00 adenylových zbytk ) na γ konci. Polyů konec je d leţitý pro ukončení transkripce, ídí transport globinové mrnů do cytoplazmy a chrání ji p ed endonukleázami. Takto upravená mrnů se posouvá do cytoplazmy na ribozomy, kde dochází k translaci, a p sobí jako p edloha pro vhodné aminokyseliny z trna. Poznání struktury lidských globinových gen ukázalo, ţe je u všech lidských globinových gen podobná. Kódující oblast je tvo ena γ exony, které jsou od sebe 16
18 odděleny dvěma introny, tzv. IVS-I a IVS-II. Na rozhraní exon a intron se nacházejí konvenční sest ihové nukleotidové sekvence umoţňující vazbu faktoru, které zabezpečují správné vyst iţení intron p i posttranskripční modifikaci globinové RNů. Introny se nep episují do globinové mrnů. Sekvence d leţité pro genovou expresi, tzv. konzervativní sekvence, se nacházejí v 5 promotorové oblasti a γ nep ekládané oblasti (UTR, untranslated region). V promotorech se nacházejí sekvence, pozitivně ovlivňující genovou expresi v poloze cis: TůTů box, CůůT box, duplikovaný CůCCC box (nebo invertovaný motiv GGGTG) a GůTů box, na které se váţí proteiny, které ídí transkripci. V globinovém promotoru se k němu váţe erytroidní regulační protein EKδF (Erythroid Krüppel-like factor). Tento transkripční faktor je nezbytný pro globinovou expresi a také reguluje expresi globinového represoru v erytroidních buňkách v dospělosti proteinu BCL11A. EKδF a BCδ11ů pat í mezi faktory hemoglobinového p epínání (hemoglobin switching) z HbF na HbA (Nienhuis et Stamatoyannopoulos, 1978). Dále proti směru exprese genu se nachází negativní regulační oblast (ůt) x -T y repetice, ke které se váţí negativně p sobící faktory v poloze trans. Ve γ UTR globinových gen se nachází signál pro polyadenylaci globinové mrnů. Na 5 konci globinové rodiny se nachází tzv. locus control region (δcr) tvo ený pěti hypersenzitivními místy s velkým počtem navazujících míst pro transkripční faktory nezbytné pro postupnou expresi všech gen globinové rodiny. Delece této oblasti vede k zastavení exprese globinových gen celého lokusu (Indrák, 1řřγ) Druhy hemoglobinu Všechny lidské hemoglobinové molekuly jsou tetramery sloţené ze dvou r zných pár globinových etěz. Globin se skládá ze dvou α (nebo ) etězc a dvou non α (,, nebo ) etězc. Během fyziologického vývoje se u lidí nachází tyto typy hemoglobinu (Obr. 4): - Embryonální hemoglobiny (Gower I, Gower II, Portland) - Fetální hemoglobin (HbF) - Hemoglobin dospělého typu (HbA a HbA 2 ) 17
19 Obr. 4: Syntéza globinových etězc u lidského plodu a kojence. Jednotlivé globinové etězce syntetizované ve ţloutkovém váčku, játrech, slezině a kostní d eni během ontogenetického vývoje, vyjád ené v procentu celkového Hb. P epnutí z embryonálních Hb na HbF je ukončeno v 10. týdnu; p epnutí z HbF na HbA dospělého typu probíhá v období těsně po narození (Nussbaum et al., 2004). Embryonální hemoglobin se tvo í v nezralých erytrocytech ţloutkového váčku a p etrvává v lidském embryu během prvního trimestru gravidity. V embryonálním období jsou hemoglobiny tvo eny kombinací (zeta) a (epsilon) nebo (gamma) globinových etězc. Gower I je tvo en β a β polypeptidovými globinovými etězci, Gower II tvo í β α a β polypeptidové globinové etězce a Portland je tvo en β a β polypeptidovými globinovými etězci. V těchto hemoglobinech etězce odpovídají budoucím α etězc m fetálního hemoglobinu a hemoglobinu dospělého typu, etězce jsou obdobou, a etězce. Nejvíc zastoupen (aţ 60 %) v embryonálních erytrocytech je Gower II. Fetální hemoglobin je dominantním hemoglobinem plodu. εá β α a β etězce. etězec má stejně jako etězec 1δ6 aminokyselin. G gen produkuje etězec, který má v pozici 1γ6 aminokyselinu glycin, a ů gen produkuje etězec s aminokyselinou alanin na této pozici (Indrák et al.,1992). HbF má zvýšenou afinitu ke kyslíku, zvýšenou odolnost proti alkáliím, pomalejší p eměnu na denaturovaný hemoglobin a vykazuje pomalejší elektroforetickou pohyblivost ve srování s HbA. Krvinky novorozence obsahují % HbF a jen % HbA. V 5-7. měsíci poklesne hodnota HbF pod 2 %. 18
20 Hemoglobin dospělého typu Hbů je sloţen z β α a β globinových etězc, HbA 2 tvo í β α a β etězce. Hbů tvo í asi řř % hemoglobinového spektra, mnoţství HbA 2 se během prvních rok ţivota postupně zvyšuje, aţ dosáhne hodnoty do γ,0 % celkového mnoţství hemoglobinu dospělého jedince. Dalším podtypem normálního HbA je tzv. glykovaný hemoglobin (Hbů1c). Tvorba Hbů1c vzniká v organismu zvýšenou glykosylací. Tvorba HbA1c je pomalá a ireverzibilní, závisí na koncentraci glukózy v organismu. Hbů1c dává nep ímou informaci o pr měrné hladině cukru v krvi (glykémie) v časovém období, které odpovídá biologickému poločasu p eţívání červených krvinek, má tedy velký diagnostický význam. U zdravého člověka netrpícího diabetem se hodnoty Hbů1c pohybují v rozmezí β,ř-4,0 %. Někdy se k dospělým hemoglobin m adí také Hbů 3 p edstavuje stárnoucí hemoglobin. Na etězce se váţe glutathion, tento hemoglobin má menší elektroforetickou pohyblivost. 19
21 2. Hemoglobinopatie Hemoglobinopatie jsou nejrozší enější monogenně dědičné choroby na světě postihující p ibliţně 7 % světové populace (Weatherall, 2001). Lze je rozdělit do dvou základních skupin podle toho, zda vznikají strukturálně abnormální globinové etězce v d sledku záměny aminokyseliny v globinovém etězci - hemoglobinové varianty (kvalitativní poruchy), nebo zda dochází k poruše tvorby některého z globin, čímţ vznikne nerovnováha v poměru α a non α globinových etězc v erytrocytech - talasemie (kvantitativní poruchy) (Lukens, 1999 Clark &Thein, 2004 Thein 2004 Wheatherall, 2006 Wheatherall, 2010). Další skupina dědičných globinových poruch je zp sobena poruchou p epínání exprese globinových etězc, ke kterému dochází v pr běhu ontogeneze. Jedná se o poruchy p echodu ze syntézy HbF na Hbů, tzv. dědičné přetrvávání produkce fetálního hemoglobinu (HPFH) (Clegg et al., 1979) Strukturální hemoglobinové varianty Strukturální hemoglobinové varianty neboli abnormální hemoglobiny vznikají v d sledku odlišné primární struktury, jejíţ p íčinou na genové úrovni jsou nejčastěji bodové mutace v kódujících oblastech globinových gen, ale mohou to být i krátké delece, elongace či genové fúze. Funkce a stabilita a tím i klinické projevy mutantních hemoglobin p edstavují široké spektrum fenotypických projev, od normálních bez klinické manifestace aţ po výrazně pozměněné, zp sobující typické hematologické syndromy. Dosud bylo popsáno více neţ 1000 hemoglobinových variant (aktualizace na Globin Gene Server, Většina z nich jsou varianty beta nebo alfa globinového etězce, vzácnější jsou varianty delta a gamma globinových etězc a hybridní globiny (produkty fúzních gen ). V České republice jsou hemoglobinové varianty vzácné (Divoký et al., 2005). Strukturální varianty se podle p evládající změny funkce rozdělují dále na a) hemoglobin S aj. hemoglobiny vedoucí ke zkrácení doby p eţívání erytrocytu (autosomálně recesivní), b) nestabilní hemoglobiny (autosomálně dominatní), c) hemoglobiny se změněnou afinitou ke kyslíku (autosomálně dominantní) a d) hemoglobiny M (autosomálně dominantní). 20
22 Hemoglobin S (HbS, 6Glu Val) zp sobuje tzv. hemoglobinopatii S označovanou rovněţ jako srpkovitá anémie (podle specifického srpkovitého tvaru erytrocytu), která postihuje zejména homozygoty a vyskytuje se p eváţně v černošské populaci v malarických oblastech. Záměnou polární a hydrofilní aminokyseliny za nepolární a hydrofobní dochází k sníţení rozpustnosti deoxygenovaného HbS v porovnání s HbA a k jeho polymerizaci, jejímţ následkem vzniká srpkovitý tvar erytrocytu. Hemoglobin E (HbE, 26Glu Lys) je pravděpodobně celosvětově nejrozší enějším strukturně abnormálním hemoglobinem, vyskytuje se zejména v jihovýchodní ůsii (Nussbaum et al, 2004). ůminokyselinová substituce aktivuje skryté 5 sest ihové místo (GTG/GTGAGG), které je lokalizováno v kodónu 25, a zp sobuje alternativní sest ih části mrnů (Indrák, 1řřγ). Tvo í se tedy normální i patologická mrnů, která je nestabilní a p edstavuje γ0-35 % hemoglobinu. V d sledku sníţené produkce E etězc vytvá í mírný talasemický fenotyp (podobným fenotypem se vyznačuje i Hb Knossos a Hb Malay (Yaish, 2013)). Hemoglobin C (HbC, 6Glu δys) je p íčinou t etí nejčastější hemoglobinopatie, hemoglobinopatie C. ůminokyselinovou záměnou vzniká méně rozpustný hemoglobin, který v deoxygenované formě vytvá í krystalky, čímţ dochází k zhoršení deformovatelnosti erytrocytu. Tyto strukturní varianty hemoglobinu jsou většinou snadno prokazatelné elektroforézou hemoglobin. Nestabilní hemoglobiny mají niţší stabilitu neţ normální hemoglobin vlivem nap. aminokyselinové záměny v blízkosti hemové kapsy narušující tak vazbu hemu na globin nebo ovlivňující kontakt mezi jednotlivými podjednotkami. Tyto hemoglobiny snadno podléhají denaturaci a precipitaci ve formě tzv. Heinzových tělísek. Ve většině p ípad dochází p eváţně k poškození globinového etězce. V České republice jsou známé unikátní varianty Hb Hradec Králové a Hb Haná, nejčastější je Hb Köln (Divoký et al., 2005). Hemoglobiny s odlišnou afinitou ke kyslíku mohou mít afinitu a) zvýšenou, čímţ dochází ke sníţenému uvolňování kyslíku ve tkáních a tkáňové hypoxii (pat í sem i Hb Olomouc) nebo b) sníţenou vedoucí k sníţené produkci erytropoietinu a lehké anémii, někdy cyanóze. Hemoglobiny M jsou p íčinou familiární cyanózy, hereditární methemoglobinémie (viz kapitola 4.2.). 21
23 Některé abnormální hemoglobiny se projevují jako talasemie. Jde o jednoduché mutace, které současně vytvá ejí abnormální globinový etězec, dále vyvolávají i poruchy v transkripci HBB genu. Tzv. talasemické hemoglobinopatie se dělí podle defektu, který vyvolává talasemický projev a to zda dochází k a) vytvo ení hybridního genu (Hb Lepore), b) ztrátě normálního terminátoru (Hb Constant Spring prodlouţený etězec α, častý výskyt v jihovýchodní ůsii) a c) aktivaci sest ihových míst (HbE, který má talasemický rys (Brabec, 1988). Vzhledem k tomu, ţe je práce zamě ená na talasemie budou kvantitativní globinové poruchy talasemie blíţe popsány v samostatné kapitole (kap. 3.) Hereditární perzistence fetálního hemoglobinu Hereditární perzistence fetálního hemoglobinu F (HPFH, hereditary persistence of fetal hemoglobin) je charakterizována p etrváváním exprese globinových gen v dospělosti zp sobené narušením mechanismu perinatálního p epnutí syntézy z na globin. Homozygoti i heterozygoti mají zvýšenou hladinu HbF homozygoti mívají některé znaky talasemií, ale nemají anémii (Indrák, 1řřγ). Za normálních okolností tvo í HbF v dospělosti méně neţ 1 % celkového mnoţství hemoglobinu. Zvýšení HbF bývá spojeno s některými talasemickými mutacemi nebo s talasemickými delecemi. Podle molekulárně genetické p íčiny rozeznáváme deleční a nedeleční formy HPFH. Pro deleční formu je charakteristická pancelulární, rovnoměrná distribuce HbF ve všech erytroidních prekurorech. Tato forma je zp sobena několika rozsáhlými delecemi, které postihují a globinové geny, nebo zvýšenou expresí G genu nebo obou gen G i A. Hladina HbF je u hetezygot v rozmezí % a u homozygot 100 % HbF. Nedeleční dědičné p etrvávání produkce fetálního hemoglobinu je zp sobeno bodovými mutacemi v promotorové oblasti jednoho nebo obou globinových gen. εutace většinou zvyšují afinitu vazebných míst pro pozitivní transkripční faktory. Odhalení p íčin HPFH se provádí molekulárně-genetickými metodami ve specializovaných laborato ích. 22
24 3. Talasemie Slovo talasemie je eckého p vodu, odvozeno z eckého slova thalassa, které znamená mo e, a emia tj. související s krví. Talasemie jsou autosomálně, většinou recesivně dědičné choroby vyskytující se zejména v oblastech εediteránu, ůfriky a ůsie. Jsou zp sobené nerovnováhou α a non-α globinových etězc, jejíţ míra se odráţí v klinickém projevu talasemií. Obrovská heterogenita talasemických alel svědčí pro navzájem nezávislý, lokální p vod těchto mutací (Weatherall et al., 2001; Steinberg et al., β001). Díky zvýšené migraci obyvatelstva se některé talasemické alely vyskytují i ve st ední Evropě, některé mutace zde vznikly de novo, avšak díky nep ítomnosti selekčního tlaku malárie je jejich rozší ení izolované. Postihují jen několik rodin a vesměs se jedná o heterozygoty, nosiče talasemické nebo strukturní hemoglobinové mutace (Divoký et al., 2005). Podle klinického a laboratorního obrazu dělíme talasemie na α talasemie, talasemie a talasemie talasemie ůlfa talasemie jsou charakterizované kvantitativní poruchou v tvorbě α etězc. Dědičné poruchy tvorby α globin ovlivňují vznik fetálního i dospělého hemoglobinu, a tudíţ zp sobují onemocnění jak v intrauterinním, tak i v postnatálním období (Nussbaum et al., 2004). ůlfa genový komplex s α1 a αβ geny (HBA1 a HBA2) se nachází na chromozomu 16 (kap. 2.3.). U α talasemií rozeznáváme β základní fenotypy α 0 talasemie (kompletně chybí syntéza α etězc inaktivací obou α gen ) a α + (částečně redukována syntéza α etězc, kdy je deletován nebo inaktivován pouze jeden α gen). εezi světově nejrozší enější delece pat í αγ.7 (delece úseku DNů o velikosti γ,7 kb) a αδ.β (delece δ,2 kb DNA). Podle klinických projev pak rozlišujeme δ skupiny α talasemií: 1. Tiché nosičství nastává p i poruše jednoho α globinového genu, genotyp (-α/αα) Jediným p íznakem je mikrocytóza bez anémie. Prokázat se dají p i molekulárněgenetickém vyšet ení. 23
25 2. Alfa talasemie minor nastává p i ztrátě funkce dvou α gen, genotyp (--/αα) nebo (-α/-α). Klinicky se projevuje mikrocytózou se zvýšeným počtem erytrocyt a nerovnováhou v syntéze α a etězc (Indrák, 1řřγ). 3. Choroba hemoglobinu H (HbH) ( 4) vzniká p i deleci γ α globinových gen, nejčastější genotyp (--/-α). HbH, který je nestabilní, jeho intracelulární precipitace zp sobuje zkrácené p eţívání erytrocyt s Hb mezi g/l. V prvním roce ţivota dochází k rozvoji anémie, vzniká ţloutenka hemolytického charakteru a hepatosplenomegalie. V krevním obraze je patrná st edně těţká anémie s MCV 70 fl. Nátěry periferní krve ukazují hypochromii, mikrocytózu, anizocytózu, terčovité erytrocyty a bazofilní tečkování. HbH lze prokázat barvením brilantní krezylovou mod í, kdy dochází ke vzniku inkluzí nestabilního HbH (Penka et al., 2001). 4. Syndrom hemoglobinu Bart s ( 4) vzniká p i deleci všech čty α globinových gen, genotyp (--/--). Jde o nejzávaţnější formu homozygotní α 0 talasemie vyskytující se p eváţně v jihovýchodní ůsii (Nussbaum et al., 2004). ůlfa etězce se v bec netvo í, nem ţe tedy vzniknout ţádný z hemoglobin Hbů, Hbů 2, ani HbF. Vznikající hemoglobin Bart s má vysokou afinitu ke kyslíku a d sledkem je nedostatečné p edávání kyslíku do tkání. Tento stav je neslučitelný se ţivotem. Většinou dochází k úmrtí plodu ve βδ. aţ γδ. týdnu nitroděloţního ţivota (tzv. hydrops fetalis) (Penka, β001). Pokud se dítě narodí má velké procento hemoglobinu Bart s a % hemoglobinu Portland (Wheatherall et al., 2006). ůlfa talasemické alely se vyskytují hlavně na západním pob eţí ůfriky a jihovýchodní ůsii. ůţ Ř0 % populace v Papui Nové Guinei jsou nosiči deleční formy a talasemie (Weatherall et al., 2006), v Saudské ůrábii má aţ 50 % obyvatel klinicky němou formu α talasemie a roste zde i počet nemocných s hemoglobinem H, v Thajsku má mutaci alfa-talasemického genu aţ 10 % obyvatelstva. V Severní ůmerice se vyskytuje alfa talasemie u černošského obyvatelstva (3 %), ale většinou ve formě minor (Fábryová, 2007). Nemocní s α talasemií se vyskytují i v České republice. Je t eba na ní myslet u mikrocytární anémie po p edchozím vyloučení sideropenie a talasemie. Pro pr kaz delecí v α globinovém lokusu se vyuţívá genového mapování nebo metod PCR HbH je prokazatelný elektroforézou. 24
26 3.2. talasemie Beta talasemie jsou charakterizovány dědičnou poruchou syntézy globinového etězce. Beta globinový komplex se nachází se na 11. chromozómu (kap. 2.3.). Nejčastějšími mechanismy zodpovědnými za talasemie, na rozdíl od α talasemií kde p evaţují delece, jsou bodové mutace globinového genu nebo jeho promotoru, které sniţují produkci funkční globinové mrnů a tím syntézu normálního genu pouze malé procento talasemií je zp sobeno delecemi části nebo celého globinového genu. Pokud vedle patologické prekurzorové mrnů vzniká i normální prekurzorová mrnů, mluvíme o + talasemii (defekt jednoho genu). Zablokuje-li mutace vznik veškeré normální globinové mrnů, mluvíme o 0 talasemii (postiţení obou gen homozygoti, dvojití heterozygoti). Na molekulární úrovni jsou talasemie mimo ádně heterogenní (Obr. 5). V souvislosti s -talasemickým fenotypem bylo objeveno p es β00 mutací většinou s částečně rozdílným klinickým obrazem (Yaish, 2013). Obr. 5: P íklady bodových mutací globinového genu zp sobujících talasemii. Upraveno dle Hoffbranda a Pettita, Většina mutací vede k autosomálně recesivnímu onemocnění (heterozygot je asymptomatický) kromě malé skupiny mutací, které jsou autosomálního dominantně 25
27 dědičného charakteru (heterozygot má projevy -talasemie intermedia) a vznikají většinou v 3. exonu globinového genu. V tomto p ípadě se hovo í o tzv. dominantně dědičné talasemii s inkluzními tělísky. První mutace tohoto typu v České republice popsali ve svých pracích Indrák (1994) a Divoký (1993). Jedná se o mutaci v kodónu 11β (G T), která vytvá í p edčasný STOP kodón a vede k translaci beta etězce se 112 aminokyselinami namísto obvyklých 1δ6, dále pak mutaci 115 (C A), která vede k záměně aminokyseliny alanin za kyselinu asparagovou a tím ke vzniku nestabilní varianty hemoglobinu (Hb Hradec Králové). talasemické mutace se dělí na: 1. mutace ovlivňující transkripci globinové mrna a) mutace v promotorové oblasti 5 globinového genu - většinou se jedná o nukleotidové substituce v TůTů boxu nebo ůcůccc boxech zp sobující + talasemiie (Indrák, 1řřγ). Tyto mutace oslabují vazbu RNů-polymerázy resp. transkripčních faktor, tím sniţují transkripci globinové mrnů na β0-γ0 % normálu. Βeta globinová syntéza však stačí na to, aby u homozygot nebo dvojitých heterozygot nevznikla těţká forma talasémie. b) mutace tvořící předčasný terminační (STOP) kodon jedná se buď o nesmyslnou jednonukleotidovou mutaci nebo posunovou mutaci (inzerce, delece). Dochází k p edčasnému ukončení syntézy globinového etězce, vzniká tak 0 fenotyp. Koncentrace defektní globinové mrnů v cytoplazmě je nízká. Pravděpodobně je to zp sobeno změněným metabolismem nukleotid nebo poruchou transportu mutované globinové mrnů z jádra do cytoplazmy. Nejznámější z těchto mutací je nesmyslná mutace v kodónu 39 (CAG TůG), která se vyskytuje asi u γ0 % talasemií v ecku a Itálii a je také p evaţující talasemickou mutací na Sardínii a v δibanonu (Fábryová, β007). Je také t etí nejrozší enější talasemickou mutací v české populaci (Divoký et al., 2005). 2. mutace poškozující zpracování mrna a) mutace v oblasti konvenčních nukleotidových sekvencí sestřihu - jedná se o mutace v 5 donorových místech sest ihu, které mají sekvenci GT a v γ akceptorových místech se sekvencí ůg. Tyto sekvence jsou součástí tzv. konvenčních nukleotidových sekvencí p sobících jako signály pro sest ih primárního transkriptu 26
28 (Indrák, 1řřγ). Tyto mutace brání normálnímu sest ihu a jsou provázeny vznikem 0 talasemického fenotypu. Výsledkem je absence funkční mrnů. Do této skupiny pat í i nejčastější mutace v české populaci, záměna adeninu za guanin v akceptorovém místě sest ihu 1. intronu globinového genu IVS-I-1. Studium haplotyp nosič této mutace v naší populaci ukázalo na pravděpodobný mediteránní p vod (Divoký et al., 2005). εutace zasahující sekvence bezprost edně sousedící s místy sest ihu mohou také ovlivnit sest ih, dále také mohou odkrýt kryptická místa sest ihu, která normálně nefungují, a m ţe se tvo it nová abnormální mrnů. Posun v místech sest ihu redukuje účinnost sest ihu, v těchto p ípadech vzniká + talasemie. b) mutace tvořící nové signály sestřihu - substituce nukleotid uvnit intronu m ţe vytvo it nové donorové nebo akceptorové místo sest ihu, které však nebrání vyuţití normální sest ihové cesty. Nap íklad substituce G A v pozici 110 v IVS-1, která je jedna z nejčastějších mutací v oblasti St edomo í, vytvá í nové sest ihové místo, které p ipomíná normální akceptor a které je p ednostně vyuţíváno (jen asi 10 % sest ihu se uskuteční v normálním sest ihovém místě (Wheatherall et al., 2006)). Tato mutace vede k sníţené produkci mrnů. Podobně je na tom mutace, která vytvá í nové akceptorové místo v pozici 116 v IVS-I a vede k malé nebo ţádné produkci mrnů a k fenotypu 0. Několik mutací, které vytvá ejí nová donorová místa, bylo popsáno také v IVS-II (Weatherall et al., 2006). c) mutace způsobující aktivaci kryptických míst sestřihu - v kryptických sest ihových místech exon m ţe nukleotidová substituce alternovat sekvenci, která je podobná donorovému sest ihovému místu, ale pro sest ih normální mrnů se neuţívá. Pokud vznikne tato sekvence blízko konvenční nukleotidové sekvence pro donorové sest ihové místo je kryptické místo aktivováno a jeho vyuţití jako donorového místa sest ihu vede k tvorbě abnormální mrnů a k poklesu tvorby normální mrnů. Takové mutace mohou být současně i p íčinou aminokyselinové záměny (Indrák, 1řřγ). Fenotypovým projevem je + talasemie nebo hemoglobinopatie. Tuto poruchu najdeme u talasemie E, talasemie Knossos a talasemie εalay. d) mutace způsobující defekt polyadenylace RNA - šest nukleotid za koncem t etího exonu (ůůtůůů) spouští enzymatický proces, který zastavuje transkripci mrnů a uvolňuje ji k dalšímu zpracování. Substituce bází, které změní tento signál, vede 27
29 k transkripci aţ k dalšímu o ř00 nukleotid vzdálenějšímu polyadenylačnímu místu. Tím vzniká dvojnásobně dlouhá mrnů, která je nestabilní (Fábryová, β007). e) mutace v oblasti čepičky - substituce C A na první pozici v této oblasti sniţují velikost transkripce, p ičemţ destabilizuji mrnů. Zp sobují + talasemii. f) mutace spojené s delečními defekty bylo popsáno minimálně 17 delečních mutací ovlivňujících beta globinový gen (Weatherall et al., β006). Nejrozší enější je delece 61ř bp nukleotid, která je rozší ena hlavně v Indii a Pákistánu, kde p edstavuje aţ 50 % talasemií (Wheatherall et al., 2006) zasahuje γ konec globinového genu, ale 5 konec z stává nedotčen. Tyto delece začínají v druhém intronu a sahají aţ za γ konec globinového genu. Velkými delecemi několika kilobází DNů jsou většinou deletovány i geny eventuálně i geny genového komplexu a vznikají, talasemie nebo syndrom dědičného p etrvávání fetálního hemoglobinu (Indrák, 1řřγ). Delece spojené se ztrátou části 5 konce globinového genu jsou spojené se zvýšenou koncentrací Hbů 2 a HbF. Do této skupiny mutací pat í i mutace československého p vod na 5 konci. Jedná se o deleci 4237 bp (Popovich et al., 1986). Zajímavým mechanismem vzniku talasemie je somatická delece globinového genu v rámci hematopoézy, která byla popsána u heterozygota pro talasemii s mozaikou buněk buď s jedním nebo ţádným globinovým genem. ůlelu pro talasemii zdědil pacient od svého otce. Během nitroděloţního vývoje vznikla delece normální mate ské alely v jednom z raných prekurzor hematopoetické vývojové ady. To vedlo k homozygozitě a k mozaicismu u krvetvorných buněk. Exprese funkčního globinového genu tak nastala jen v části erytrocyt, u pacienta se tak vyvinula talasemie intermedia (Badens et al., 2002) Klinická klasifikace talasemií Zjednodušeně a za p edpokladu normální struktury a funkce α gen m ţeme klinické projevy talasemií rozdělit do 3 skupin: 1. talasemie major Talasemie major (homozygotní nebo dvojitě heterozygotní forma 0 talasemické mutace, Cooleyʼs anemia) se vyskytuje u nemocných, kde oba geny jsou postiţeny 0 mutací nebo jeden gen je postiţen 0 a druhý gen těţkou + mutací. Jde o nejzávaţnější 28
30 formu talasemie, která je charakterizována těţkou anémií (p i dg. β0-30 g/l) a závislostí na transfúzích. Pacienti umírají ve t etí dekádě ţivota na projevy p etíţení organismu ţelezem v d sledku transfúzí. Onemocnění bývá obvykle diagnostikováno po uplynutí prvních δ týdn ţivota. ůsi v 6. týdnu se vyvíjí anemie s p ítomností jaderných buněk v periferní krvi, v Ř. týdnu splenomegalie. Dítě má nízkou postavu s velkou hlavou s vyčnívajícími lícními kostmi, širokým nosem a oddalujícími se horními zuby. V dalším vývoji dochází k retardaci r stu, postiţení sexuálního vývoje a postiţení endokrinních tkání (diabetes mellitus, hypoparatyroidismus aj.). Díky vystupňované erytropoéze dochází k rozši ování d eňové dutiny a ztenčování kompaktní kosti nejčastěji jsou změny patrné na rukou a chodidlech, dále na lebce hlavy. Postupně dochází k postiţení ostatních orgánových systém hepatobiliárního traktu (nejprve extramedulární hematopoéza, postupně cirhóza), kardiovaskulární komplikace p edevším v souvislosti s p etíţením ţelezem, se objevují jiţ v první dekádě ţivota. Na p ítomné splenomegálii se podílí, mino jiné i extramedulární hematopoéza (Penka, β001). V krevním obraze dominuje těţká mikrocytární hypochromní anémie s hodnotami hemoglobinu pod 70 g/l se z etelnou anizocytózou (red blood cell distribution width/distribuční ší e erytrocyt, RDW nad 15,2). V nátěrech je patrná kromě anizocytózy i poikilocytóza, terčovité erytrocyty, bazofilní tečkování. Hodnoty HbF a HbA 2 jsou výrazně zvýšeny a v dospělosti je prakticky eliminován HbA. V léčbě se uplatňují δ hlavní léčebné postupy substituční léčba, odstraňování nadbytečného ţeleza chelatačními látkami, splenektomie a transplantace kostní d eně (Penka, 2001). V České republice je talasemie major diagnostikována výjimečně u potomk imigrant z malarických oblastí. 2. talasemie intermedia Talasemie intermedia vzniká p i postiţení jednoho genu 0 mutací a druhého genu mírnou formou + mutace, nebo p i postiţení obou gen + mutací. Vyznačuje se obecně méně závaţnou anémií s občasnou pot ebou transfuzí a p etíţení ţelezem není tak závaţným problémem. Nicméně klinické projevy se mohou výrazně lišit, od asymptomatických (Hb mezi 100-1β0 g/l) na úrovni talasemie minor aţ po těţké formy s retardací r stu a splenomegalii (Hb kolem 60 g/l) jako v p ípadě talasemie major. Kostní d en je hyperplastická. V normoblastech je moţně prokázat inkluze z denaturovaných α 29
31 etězc. Hladina Hbů je obvykle větší neţ γ0 % a hladina HbF menší neţ 70 %. Pacienti se doţívají dospělého věku. Výskyt v České republice je vzácný. 3. talasemie minor Talasemie minor (heterozygotní) je nejčastější, klinicky němou, formou talasemie. Vzniká p i postiţení jen jednoho z gen 0 nebo + mutací. Záchyt nemocných, kte í jsou klinicky asymptomatičtí, je spíše náhodný. εají mírnou anémii (Hb kolem g/l) s nevýraznou retikulocytózou, ale s výraznou mikrocytózou (εcv fl) a erytrocytózou (Penka, β001). ůnémie se m ţe prohloubit během gravidity. V krevních nátěrech jsou patrné i terčovité erytrocyty, hypochromázie a bazofilní tečkování. Většinou se nachází zvýšená hladina Hbů 2 (3,5-7,0 %) i hladina HbF (do 6,0 %). Je zde lehká hyperplazie erytropoézy se zvýšenými zásobami ţeleza. Výskyt v České republice je relativně častý Patofyziologie talasemií U homozygot s talasemií deficit etězc zp sobuje akumulaci α etězc. Vzhledem k jejich velké nestabilitě volné α etězce agregují a vytvá í nerozpustné inkluze v erytroidních prekurzorech kostní d eně. Na rozdíl od Heinzových tělísek, která vznikají z denaturovaného hemoglobinu, jsou talasemické inkluze sloţené jen z α etězc. Část inkluzí mohou p edstavovat i hemichromy. Inkluze tvo í velkou masu, která znemoţňuje další vývoj prekurzor v kostní d eni, a dochází k inefektivní erytropoéze. Inkluzní tělísko narušuje plasticitu erytrocytární membrány a zhoršuje pr chodnost erytrocyt mikrocirkulací zvláště ve slezině, coţ vede k deformaci a rozpadu červené krvinky (Divoký et al., 2005). V homozygotním stavu uniká destrukci jen asi % normoblast. Inefektivní erytropoézou dochází rovněţ ke sníţení hladiny hepcidinu (viz kap. 5.3) a zvýšené absorbci ţeleza, k jehoţ akumulaci p ispívá rovněţ trasfúzní léčba, coţ vede ve finále k přetížení organismu železem. Vlivem poruchy v tvorbě globinových etězc je sníţená rovněţ inkorporace ţeleza do porfyrinogenu vedoucí k menší produkci hemových molekul. Volné ţelezo tak agreguje ve formě feritinu nebo hemosiderinu v cytoplazmě a mitochondriích. Zvýšené mnoţství ţeleza se nachází i extracelulárně. 30
32 Ţelezo samo o sobě p sobí jako oxidační činidlo a m ţe vést k peroxidaci lipid a bílkovin a sniţovat tak ţivotaschopnost talasemických erytrocyt, které jsou tak vnímavější na oxidační stres. Inefektivní erytropoéza vede k zesílení kompenzační erytropoézy. ε ţe docházet k poškození kostní struktury, ztenčení kompaktní tkáně a rozší ení medulární dutiny. Někdy m ţe d eň proniknout z d eňové dutiny ven a vytvá í extraoseální (periférní) masy. Extramedulární hemopoéza m ţe probíhat i ve slezině a játrech. V literatu e je zmiňována rovněţ zvýšená agregabilita erytrocyt a jejich adherence k endotelu vedoucí k mikrocirkulačním změnám. ůdherence je vysvětlována zvýšenou expozicí fosfatidylserinu na povrchu membrány erytrocytu a zvýšenou expresivitou molekuly CDγ6, která ulehčuje interakci mezi erytrocytem a endotelem (Fábryová, β007) Diagnostika Většina jedinc s talasemií v české a slovenské populaci jsou heterozygoti. Tito nosiči talasemické alely mívají asi dvojnásobně zvýšenou hladinu Hbů 2 oproti normálu (na 3,5-7,0 %) a asi 1/γ nosič má i zvýšenou hladinu HbF. Nejrozší enějším diagnostickým testem je tak chromatografické nebo elektroforetické stanovení hladiny HbA 2 a stanovení HbF alkalickou denaturací. P ímá detekce mutací na DNů úrovni vyuţívá molekulárně-genetických metod: restrikční analýzu, PCR a sekvenování. Tyto metody jsou rychlé a spolehlivé a jsou vyuţívány i v rámci prenatální diagnostiky (Divoký et al. 2013) Výskyt talasemií Výskyt beta talasemických alel je stejně tak jako u většiny hemoglobinopatií je soust eděn do malarických oblastí (HbF je rezistentní k natrávení hemoglobinázami produkovanými Plasmodium falciparum a brání tak r stu tohoto parazita (Shear et al., 1998). V Evropě je ohniskem výskytu hlavně Itálie, dále ecko, Sardinie, Sicílie a Kypr (Cao et al., 2001). Beta talasémie se vyskytuje rovněţ v západní ůfrice, Turecku, Iránu, Sýrii, v Saudské ůrábii a v Pákistánu (Weatherall et al., 2006). V Severní ůmerice se beta talasemie nejvíce vyskytuje mezi italskými a eckými p istěhovalci. V jihovýchodní/východní ůsii p evaţuje výskyt α talasemické alely. 31
33 V České a Slovenské republice je talasemie (a to zejména talasemie minor pop. intermedia) nejčastější p íčinou vrozené mikrocytární anemie a to i p esto, ţe je jejich výskyt ve st ední Evropě relativně vzácný. Podobně jako v jiných nemalarických oblastech zastoupení talasemie odráţí historickou i současnou migraci a demografické změny, které ovlivňují genetickou variabilitu nynější populace ţijící v této oblasti (Hlobilová, 2011) talasemie a talasemie δ talasemie jsou relativně vzácná skupina chorob s klinicky mírnými p íznaky zp sobenými chyběním a globinové produkce, která je nahrazena syntézou etězc (Indrák, 1řřγ). εolekulárně genetickým podkladem těchto chorob jsou většinou rozsáhlé delece DNů nebo, vzácněji, kombinace delece s bodovou mutací postihující současně i globinový gen (Indrák, 1řřγ). Homozygoti s talasemií mají relativně mírné klinické projevy a malou aţ st ední splenomegálii. Chybí HbA a HbA 2, dominuje HbF. Vznik talasemického fenotypu je podmíněn nerovnováhou v produkci α a globinových etězc (γ:1 ve prospěch α etězc ). Heterozygoti mají nenápadnou mikrocytózu a hypochromázii s normální hladinou Hbů 2 a se zvýšenou hladinou HbF (Indrák, 1řřγ). Gama globinové etězce produkují β globinové geny, které jsou uloţeny v genovém komplexu za sebou v po adí 5 -G -A -γ. Sloţení HbF se m ţe lišit v závislosti na rozsahu delece. Z delecí vedoucích k 0 talasemii je v Evropě častá tzv. sicilská 1γ kb dlouhá delece, vyskytující se vzácně i v ČR. Homozygoti produkují pouze HbF, heterozygoti mají talasemický krevní obraz s 5-15 % HbF (Divoký et al., 2005). Zvláštní skupinou delečních forem talasemíí jsou delece vznikající nerovnoměrným crossing-overem v meióze mezi a globinovými geny. Vzniká fúzní gen pod vlivem globinového promotoru. Produktem těchto fúzních gen jsou chimerické etězce, které se kombinují s α etězci a dávají vzniknout skupině Hb δepore s talasemickým fenotypem (Divoký et al. 2013). Heterozygoti mají projevy talasemie minor, homozygoti talasemie major. Známé jsou γ typy Hb δepore, které se odlišují fúzním místem a etězc : Hb Boston, Hb Lepore-Hollandia a Hb Lepore-Baltimore. Hemoglobin δepore byl dosud prokázán v české populaci u více neţ 7 rodin (Divoký et al. 2013). 32
34 δ talasemie je vzácným typem talasemie, která vzniká delecemi celého globinového komplexu (Harteveld et al., 2003). V dospělosti se projevují jako talasemie minor. Na rozdíl od talasemických heterozygot nemají zvýšenou hladinu Hbů 2. V České republice zatím nebyly tyto formy talasemie diagnostikovány. 33
35 4. Methemoglobinémie εethemoglobinémie je onemocnění charakterizované p ítomností abnormálně vysokého mnoţství methemoglobinu (methb) v krvi. εethemoglobin je oxidovaná forma hemoglobinu, která prakticky není schopna vázat kyslík. Za fyziologických podmínek nep ekračuje koncentrace methb 1 %. Zvýšení jeho koncentrace se tak m ţe projevit tkáňovou hypoxií, která m ţe být kompenzována mírnou polycytémií, a cyanózou (namodralé zbarvení/modré zabarvení k ţe a viditelných sliznic) a to p i koncentraci methb nad 50 g.l -1 (tj. cca nad 25 %). εethemoglobinémie m ţe být vrozená nebo získaná. O vrozené methemoglobinémii hovo íme zejména v souvislosti s a) deficitem enzymu cytochrom-b5-reduktázy, nebo b) p ítomností abnormálních variant hemoglobinu (hemoglobin ε). εethemoglobinémie m ţe být také asociována s deficitem pyruvátkinázy nebo glukóza-6-fosfátdehydrogenázy v d sledku narušení produkce kofaktor NůDH a NůDPH nebo s p ítomností nestabilního hemoglobinu (Percy & Lappin, β00ř). P íčinou získané methemoglobinémie (nejčastěji polékové) je expozice exogenním oxidant m a jejich metabolit m antibiotika (sulfonamidy aj.), lokální anestetika a další látky (anilinová barviva, dusitany, dusičnany). Na rozdíl od vrozené formy m ţe získaná, toxická methemoglobinémie, ohroţovat pacienta na ţivotě Deficit cytochrom-b5-reduktázy Methemoglobin je za normálních podmínek v organismu odbouráván z 99 % enzymem cytochrom-b5-reduktázou (Cb5R, EC , NADH-methemoglobinreduktáza, diaforáza) (Obr. 6) (Mansouri & Lurie, 1993). Jedná se o γ5 kda (γ01 aa) flavoprotein (FAD) (Obr. 7). εembránově vázaná varianta Cb5R (mitochondrie, endoplazmatické retikulum) stejně tak jako erytrocytární solubilní varianta je kódována genem (CYB5R3), který je lokalizován na chromosomu ββ (ββq1γ-qter) a obsahuje ř exon ; v p ípadě solubilní formy se nep episuje část exonu, protein je kratší o β5 aminokyselin. 34
 v metabolismu glykolýzy.")
, tj. methb 1 %, která byla poprvé popsána v roce 1943 (Percy & Lappin, 2008).")
36 Obr. 6: Redukce methemoglobinu na hemoglobin prost ednictvím cytochrom-b5-reduktázy (Cb5R) NůDH vzniká reakcí glyceraldehyd-3-fosfátu (G3P) na 1,γ bisfosfoglycerát (1,3-BPG) zprost edkovanou enzymem glyceraldehyd-3-fosfátdehydrogenázou (G3PD) v metabolismu glykolýzy. NůDH následně redukuje enzymový FAD a ten dále cytochrom b5, který p sobí jako elektronový donor a redukuje methemoglobin (Fe +++ -Hb) na hemoglobin (Fe ++ -Hb) (p evzato z Prehal, 2000). Nedostatek Cb5R vede k vrozené autozomálně recesivně dědičné methemoglobinémii (RCε), tj. methb 1 %, která byla poprvé popsána v roce 1943 (Percy & Lappin, 2008). Pacienti, kte í jsou jednoduchými heterozygoty, jsou obvykle asymtomatičtí. Symptomy se objeví aţ u homozygot nebo dvojitých heterozygot, u kterých většinou vzroste úroveň methb nad 25 % (Jaffe, 1986). Pokud se hladina methemoglobinu blíţí 70 %, m ţe nastat cévní kolaps, kóma a smrt (Percy & Lappin, 2008). Obr. 7: Krystalová struktura proteinu Cb5R s FAD- a NAD(P)H-vazebnou doménou (znázorněno červeně resp. mod e) a s vyobrazenou vazbou FůD prostetické skupiny a navázaného NůD + (Percy & Lappin, 2008). 35
37 Rozlišujeme β typy těchto methemoglobinémií: - Typ I (RCM typu I) - funkční deficit Cb5R postihuje jen erytrocyty RCM typu I vzniká v d sledku mutací genu CYB5R3, které neovlivňují rychlost exprese Cb5R, ale které mají vliv na stabilitu enzymu. Kromě cyanózy jsou častými vedlejšími p íznaky bolesti hlavy, únava a dušnost p i námaze. Většina jedinc je však zcela asymptomatická. - Typ II (RCM typu II) - deficit Cb5R postihuje všechny buňky RCM typu II je zp sobena mutacemi, které sniţují expresi Cb5R nebo ovlivňují celkovou aktivitu enzymu. Tento deficit se vyskytuje v % p ípad postiţených vrozenou methemoglobinémií. U RCε typu II bývá cyanóza doprovázena mentální retardací a neurologickými symptomy (Percy & Lappin 2008; Prchal & Gregg, 2005). Obvykle se projeví během prvních δ měsíc ţivota. Bylo identifikováno p es 33 r zných CYB5R3 mutací, z nichţ některé jsou společné pro oba typy RCε (Percy at al., 2002). Od těchto forem methemoglobinémie je nutné odlišit hereditární methemoglobinémii typu III spojovanou s Hb ε, která je však autosomálně dominantního charakteru, a asociované formy s jinými poruchami nebo získané formy methemoglobinémie. K léčbě methemoglobinémie se pouţívá methylenová mod nebo kys. askorbová Hemoglobin M εethemoglobinémie (familární cyanózy) zp sobené hemoglobinovými variantami, které souborně označujeme jako hemoglobiny M (Hb ε), jsou skupinou autosomálně dominantních onemocnění. Nejčastěji vznikají substitucí proximálního (pozice 92) nebo distálního (pozice 6γ) histidinu (hem-kontaktní aminokyseliny) v α nebo etězcích tyrozinem. Tyrozin svou hydroxylovou skupinou vytvá í komplex s Fe 3+ a stabilizuje ţelezo v jeho trojmocném stavu (ferrihem, Fe 3+ ). Tato forma hemu tak není schopna reverzibilně vázat kyslík navíc hemové skupiny v tetrameru hemoglobinu s dvojmocným ţelezem mají zvýšenou afinitu ke kyslíku. Známými variantami jsou Hbε-Saskatoon ( 6γ His>Tyr), popsaný i v slovenské populaci (Divoký et al., 2005), HbM-Milwaukee-I ( 67 36
38 Val>Glu), HbM-Hyde Park ( řβ His>Tyr) a Hbε Boston (α5ř His>Tyr). Pacienti jsou heterozygoti s obsahem methb kolem % homozygotní forma je neslučitelná se ţivotem. Kromě methemoglobinémie s projevem cyanózy se u nich často vyskytuje i hemolytická anémie ada Hb ε je současně nestabilních. 37
39 5. Přetížení organismu železem 5.1. Železo v lidském těle Železo (Fe) je esenciálním prvkem ţivých organism. Vyskytuje se jak ve formě vázáné na proteiny (hemové nebo nehemové ţelezo) nebo nevázané. εezi tzv. hemové proteiny, u kterých dochází k vazbě ţeleza na jejich prostetickou skupinu protoporfyrinu IX pat í hemoglobin (Ř5 % neskladovaného ţeleza) slouţící k transportu kyslíku, myoglobin, který plní d leţitou zásobní funkci kyslíku ve svalech, cytochromy zajišťující p enos elektron, a některé enzymy podílející se na metabolismu kyslíku (kataláza, peroxidáza aj.). Nehemovými proteiny váţícími ţelezo jsou nap. transferin, feritin, hemosiderin, některé oxidoredukční enzymy nebo ţelezo-sirné proteinové komplexy dýchacího etězce (Pecka, β00β). Ţelezo, které není vyuţito na tvorbu hemoglobinu nebo jiných enzymatických komplex, je v organismu skladováno v hepatocytech a makrofázích. Ţelezo v nevázané formě volné ţelezo je pro buňky potencionálně toxické; m ţe totiţ katalyzovat tvorbu volných kyslíkových radikál (Fentonova reakce), které mohou p i svém nadměrném vzniku a nedostatečném antioxidačním mechanismu poškozovat organismus (oxidační stres). Vznik toxických kyslíkových radikál je však na druhé straně nezbytný pro bakteriální funkci makrofág umoţňující nitrobuněčnou destrukci některých fagocytovaných mikroorganism. Ţelezo hraje svou roli i v imunitní odpovědi organismu. Jeho deficit negativně ovlivňuje proliferaci a diferenciaci B a Th1- lymfocyt, zatímco p etíţení ţelezem vede k dysfunkci NK buněk, sníţení cytotoxicity neutrofil a změnám poměru CDδ a CDŘ lymfocyt (Horváthová & Pospíšilová, 2010). Umoţňuje však i r st a proliferaci bakterií a nádor. Vzhledem k tomu, ţe nerovnováha ţeleza (nadbytek nebo nedostatek) m ţe vést k rozvoji r zných patologických stav, je homeostáza ţeleza p ísně regulována (kap. 5.3.) Metabolismus železa Většina ţeleza kolujícího v krevním oběhu pochází z rozpadlých erytroidních buněk, k jejichţ fagocytóze (enterofagocytóze) a lýzi dochází v makrofázích retikuloendoteliálního systému (slezina, játra, kostní d eň); jen v malé mí e je vst ebáváno potravou a to zejména ve formě organické/hemové (laktoferin, kasein) pop. anorganické. 38
40 Vst ebávání hemového ţeleza v enterocytech duodena je mnohem efektivnější neţ vst ebávání ţeleza anorganického. P edpokládá se, ţe hem získány proteolytickým štěpením nejčastěji hemoglobinu a myoglobinu v ţaludku a tenkém st evě je transportován specifickým transportérem do enterocytu. Z hemu je enzymem hemoxygenázou-1 uvolněné trojmocné ţelezo, které je dále redukováno na dvojmocné prost ednictvím duodenální cytochrom-b-reduktázy a transportováno do cytoplazmy enterocytu prost ednictvím transmembránového receptoru DMT1 ( divalent metal transporter-1 ), kde m ţe být jako zásobní vázáno na feritin (Horvathová et al., 2012 Latunde-Dada et al., 2002). ůnorganické ţelezo je vst ebáváno stejnými pochody jako hemové ţelezo po jeho uvolnění z hemu (Beard & Han, 2009). Transport ţeleznatého iontu zpět p es membránu enterocytu do oběhu je zprost edkován membránovým proteinem feroportinem, který se nachází i na hepatocytech a makrofázích retikulo-endoteliálního systému. Uvolněný ţeleznatý iont je pak zpětně oxidován účinkem oxidázy hefestinu (transmembránový protein uloţený v bazolaterální membráně enterocytu duodena) nebo plasmatickou oxidázou ceruloplasminem zpět na ţelezitý. Tato oxidace je nezbytná pro jeho vazbu na transferin (TF), který má β vazebná místa, a vzniká tak holotransferin. Za fyziologických podmínek je TF saturován p ibliţně z γ0 % (Horváthová & Pospíšilová, 2010). P i nedostatku ţeleza se zvyšuje exprese genu pro TF s následným zvýšením jeho syntézy v játrech. Celkové mnoţství plazmatického TF reprezentuje celkovou vazebnou kapacitu plazmy pro ţelezo (CVK). Do erytrocyt se ţelezo dostává prost ednictvím vazby holotransferinu na transferinový receptor 1 (TFR1) a následnou endocytózou. Z holotransferinu je trojmocné ţelezo uvolňováno acidifikací a redukováno ferrireduktázou STEůPγ ( six transmembrane epithelial antigen of the prostate γ ) a transportováno nejpravděpodobněji prost ednictvím receptor DεT1 (endozóm) a mitoferinu-1 (mitochondrie) do mitochondrií, kde je za p ispění ferrochelatázy zabudováno do protoprofyrinu IX za vzniku hemu, který je uvolňován do cytoplasmy, kde vytvá í komplexy s globinovými etězci hemoglobiny (kap. 1.2.). 39
41 5.3. Regulace homeostázy železa Homeostáza ţeleza je zabezpečována udrţováním rovnováhy mezi p íjmem, spot ebou a zásobami ţeleza jak na a) úrovni samotných buněk, tak na b) úrovni celého organismu. Neexistuje totiţ ţádný jiný regulovaný mechanismus, kterým by se organismus zbavoval nadbytečného ţeleza. Buněčná regulace spočívá v koordinaci exprese protein účastnících se metabolismu ţeleza. Nejvíce byly zatím prostudovány postranskripční mechanismy regulace p sobením cytoplazmatických protein IRP1 a IRP2 ( iron regulatory protein 1 a β ) a jejich vazbou na tzv. IRE ( iron response element ) oblasti v nep ekládaných oblastech mrnů. Nachází-li se IRE v 5 -nep ekládané oblasti mrnů dochází k zablokování translace mrnů, je-li v γ -UTR, pak se naopak mrnů stává stabilnější a dochází k její translaci. Sníţením hladiny ţeleza a následnou vazbou IRP1 a IRPβ na IRE oblasti dochází k stabilizaci mrnů pro TFR1 a zvýšení p íjmu ţeleza, naopak mrna pro L- a H-feritin a ůδůsβ je nep episována, čímţ se sníţí uskladnění ţeleza a neprobíhá ani syntéza hemu (kap. 2.3.). Regulací na úrovni celého organismu se kontroluje koncentrace ţeleza v plasmě a v extracelulárních tekutinách. Centrální postavení mají v této regulaci homeostázy ţeleza játra a jejich produkt hormon hepcidin, který funguje jako negativní regulátor uvolňování jak absorbovaného ţeleza z enterocyt duodena, tak i recyklovaného ţeleza z makrofág (Ganz, β00δ). Hepcidin se váţe na feroportin, čímţ dochází k jeho internalizaci a degradaci a následně k sníţení exportu ţeleza do krevního ečiště a naopak k jeho zvýšenému ukládání (Nemeth et al., β00δ). Dochází k celkovému sníţení hladiny ţeleza v krevní plazmě. Syntéza hepcidinu je stimulována zánětlivými procesy, zvýšeným p íjmem ţeleza a tlumená nap. v p ípadě neefektivní erytropoézy, anémie a hypoxie. Naopak jeho nadměrné hladiny u lidí jsou p íčinou sníţené st evní absorpce ţeleza a vzniku anémie z nedostatku ţeleza (Beard, β00δ). Na regulaci syntézy hepcidinu v závislosti na koncentraci ţeleza se v hepatocytech jater podílí transferinový receptor β (TFRβ), protein asociovaný s hemochromatózou typu I (HFE) nebo hemojuvelin (HJV), které indukují expresi genu pro hepcidin (HAMP), a matriptáza-β, které je negativním regulátorem exprese tohoto genu. Nezávisle na p edchozí regulaci se mohou podílet na regulaci syntézy hepcidinu rovněţ cytokiny, které vznikají p i zánětlivých procesech, jako je nap. interleukin 6, který 40
42 indukuje transkripční faktor STůTγ, který p sobí jako aktivátor exprese genu HAMP. Jedná se o patologický stav uplatňující se p i rozvoji anémií chronických chorob Primární hemochromatóza Hereditární (primární) hemochromatóza je heterogenní skupina dědičných onemocnění, pro niţ je charakteristická zvýšená resorpce ţeleza v duodenu a zvýšená hladina feritinu. P ebytečné ţelezo se akumuluje v játrech, slinivce b išní, srdci, hypofýze s postupným narušením jejich funkce. Kromě nespecifických p íznak jako je únava, hubnutí a slabost, se vyskytuje hepatomegalie, zvýšená koţní pigmentace, cukrovka (u hemochromatózy díky zbarvení pokoţky označovaný také jako tzv. bronzový diabetes). Nejčastější p íčinné mutace byly identifikovány v pěti r zných genech v HFE, TFR2, HFE2, HAMP a SLC40A1. První t i se podílejí na regulaci exprese hepcidinu, HAMP je gen pro samotný hepcidin a SLC40A1 je gen pro feroportin. Podle toho se v současné době hemochromatózy rozlišují na několik typ, kromě klasické HFE-hemochromatózy rozeznáváme skupinu tzv. non-hfe-hemochromatóz, kam pat í hemochromatóza typu β-4 (Tab. 1). Tab. 1: Rozdělení vrozených hemochromatóz (upraveno dle Horváthová & Divoký, 2013). HH Gen Dědičnost Protein Funkce HH typ 1 HFE (6p21.3) AR HFE stimulace exprese HH typ 2a HFE2 (1q21) AR HJV hepcidinu HH typ 2b HAMP AR hepcidin regulace homeostázy Fe (19q13) HH typ 3 TFR2 (7q22) AR TFR2 stimulace exprese hepcidinu HH typ 4a a 4b SLC40A1 (2q32) AD feroportin transmembránový transport Fe 2+ Pozn.: HH: hereditary hemochromatosis; ůr/ůd: autosomálně recesivní/dominantní p enos. Nejčastějším typem HH je hemochromatóza typu 1 (mutace v genu HFE) popsaná Federem et al. (1řř6). Projevuje se aţ ve st edním věku (γ0-50 let) a to zejména u muţ ; u 41
43 ţen se onemocnění objevuje většinou s prodlením ve srovnání s muţi a to vlivem pravidelných ztrát ţeleza během menstruace a p i porodu. Je popsáno p es γ0 mutací a polymorfism, z nichţ nejčastější jsou mutace CysβŘβTyr, His6γůsp a Ser65Cys. Mutace Cys282Tyr se vyskytuje v homozygotním stavu aţ u Ř0 % pacient s hemochromatózou. Frekvence výskytu mutací v České populaci 15 % (His63Asp), 3,4 % (Cys282Tyr) a 1,3 % (Ser65Cys) - odpovídá p ibliţně frekvenci jejich výskytu udávanou pro st ední Evropu (Čimburová et al., β005). HFE p sobí nejen jako modulátor hepcidinu, ale interaguje rovněţ s TFR1 a ovlivňuje tak jeho afinitu k transferinu a tím i absorpci ţeleza enterocyty. U hemochromatózy typu β (neboli juvenilní manifestace do γ0. roku ţivota) rozeznáváme β typy a to typ βa (mutace v genu HFE2) a typ 2b (mutace v genu HAMP) (Papanikolaou et al., 2004 Roetto et al., 2003, Niederkofler et al., 2005). Hemochromatóza typu γ (mutace v genu TFR2) má fenotypový projev podobný HH typu 1, ale s časnějším nástupem klinických p íznak (Camaschella et al., 2000). Transferinový receptor β na rozdíl od transferinového receptoru 1 je vysoce exprimován v játrech, není regulován ţelezem a má niţší afinitu k transferinu je povaţován za jeden ze sensor ţeleza v těle regulující tak syntézu hepcidinu. U hemochromatózy typu 4 neboli také feroportinové choroby (mutace v genu SLC40A1) rozlišujeme β skupiny a to podle mutací a jejich účinku - typ δa, kdy dochází k nesprávné membránové lokalizaci feroportinu zp sobující ztrátu jeho schopnosti transportovat ionty ţeleza, které se pak hromadí p edevším v makrofázích (Horváthová & Divoký, 2013), a typ δb, kdy dochází k rezistenci feroportinu na negativní regulaci hepcidinem a proto k nadměrnému uvolňování ţeleza z enterocyt a makrofág a projev m podobným klasické hemochromatóze. Na rozdíl od p edchozích typ je HH typu δ autosomálně dominantní (Montosi et al., 2001). Pro HH typu 1, β a γ je charakteristická nízká hladina hepcidinu, coţ vede k nadměrnému uvolňování ţeleza z enterocyt a makrofág a zvyšování hladin sérového ţeleza, feritinu a saturace transferinu (Horváthová et al., 2012). U HH typu δ p evaţuje zvýšená hladina hepcidinu (Papanikolaou et al., 2005), koncentrace ţeleza v makrofázích je vysoká, saturace transferinu je buď normální (HH typ δa) nebo zvýšená (HH typ δb). Základem léčby je odstranění nadbytečného ţeleza a to zejména krevními odběry (flebotomie). 42
44 5.5. Sekundární hemochromatóza Pokud je p íčinou p etíţení ţelezem jiné onemocnění neţ hereditární hemochromatóza typu 1 aţ δ, pak hovo íme o tzv. sekundárním p etíţení ţelezem (sekundární/získaná hemochromatóza). Nejčastější p íčinou (ať uţ získanou nebo vrozenou) jsou opakované transfuze, inefektivní erytropoéza nebo chronické onemocnění jater. adí se sem nap. talasemie, sideroblastická anémie (zvýšené ukládání ţeleza v mitochondriích erytroblast v d sledku poruchy syntézy hemu nejčastěji deficitem enzymu ůδůsβ), dyserytropoetická anémie, chronická hemolytická anémie spojená s deficitem enzymu pyruvátkinázy nebo glukóza-6-fosfátdehydrogenázy, či vrozená sférocytóza. V biochemickém obraze nalézáme zvyšující se hladiny feritinu a ţeleza v séru, stoupá saturace transferinu p i jeho současném poklesu. 43
45 6. Cíl práce Cílem mé rigorózní práce bylo: 1. Analyzovat a zhodnotit výskyt talasemických mutací u souboru nemocných z ČR a SR za azených do studie od roku 2006 (ve spolupráci s Hematoonkologickou a Dětskou klinikou FN a δf UP v Olomouci). 2. Porovnat výsledky této studie s jiţ d íve publikovanými daty o výskytu talasemických mutací v ČR a SR. 3. Na biochemické a následně na molekulární úrovni charakterizovat pacienty s methemoglobinémií. 4. Na souboru pacient s deficitem pyruvátkinázy a s projevy sekundární hemochromatózy vyloučit nebo potvrdit p ítomnost kauzální mutace genu HFE (u jednoho pacienta z tohoto souboru s hyperferitinémií provést molekulární analýzy všech gen vedoucích k primární hemochromatóze). 44
46 EXPERIMENTÁLNδ ČÁST 7. Materiál 7.1. Biologický materiál Za účelem hematologického a molekulárně genetického vyšet ení bylo pouţito ml periferní krve v K 3 EDTA pro stanovení aktivity cytochrom-b5-reduktázy pak cca ř ml periferní krve v heparinu sodném Chemikálie a roztoky Komerční kity BigDye Terminator v.γ.1 Cycle Sequencing Kit (Applied Biosystems, USA) Big Dye XTerminator Purification Kit (Applied Biosystems, UK) HotStarTaq Master Mix (Qiagen, Německo) Mini Elute Gel Extraction Kit (Qiagen, Německo) Chemikálie 2-log DNA ladder (New England Biolabs, USA) 2-merkaptoethanol (Serva, Německo) ůgarosa (Invitrogen life Technologies, Velká Británie) Bromfenolová mod (P-δab, ČR) DNA loading pufr (Qiagen, EU) DTT (Sigma-ůldrich, Německo/USů) Ethanol pro UV (Tamda, ČR) Fenol (Serva electrophoresis, Německo) Glycerol (δachner, ČR) HCl (Penta, ČR) Hydroxychinolin (Serva, Německo) Chloroform (Penta, ČR) Izopropanol (δachner, ČR) K 3 Fe(CN) 6 (Serva, Německo) KCl (Fischer Scientific, USA) 45
47 KH 2 PO 4 (Sigma-Aldrich, USA) Kyselina boritá (ůmresco, USů) εikrokrystalická celulóza (Sigma-Aldrich, USA) NůDH (Serva, Německo) Na 2 EDTA (Sigma-Aldrich, USA) Na 2 HPO 4 (Duchefa, Holandsko) NaCl (δachner, ČR) NH 4 Cl (Penta, ČR) Nuclease Free Water (Ambion, USA) Primery (Eastport, Německo) (Tab. γ aţ Tab. 8) Proteináza K (Tritirachium album) (Sigma-ůldrich, Německo/USů) SDS (Serva Electrophoresis, Německo) Tris (Promega, USA) α-celulóza (Sigma-Aldrich, USA) Laboratorní roztoky a pufry 0, 15δ ε NaCl (fyziologický roztok) 10 mm PBS (ph = 7,4): 0,137 M NaCl, 2,7 mm KCl, 8,2 mm Na 2 HPO 4, 1,8 mm KH 2 PO 4 10% SDS (w/v, ph = 7,2) LSB: 0,3 M Tris (ph= 8,9), 0,75 M DTT, 12% SDS (w/v), 0,γ% bromfenolová mod (w/v), 60% glycerol δyzační roztok: 1,5 ε NH 4 Cl, 100 mm NH 4 HCO 3, 10 mm Na 2 EDTA Roztok proteinázy K: 100 µg/ml, 1% SDS (w/v), β mε EDTů (ph Ř,0) Stabilizační roztok: β,7 mε Na 2 EDTA, 0,7 mm 2-merkaptoethanol STE pufr (ph = 7,4): 100 mm NaCl, 50 mm tris (ph = 8), 1 mm EDTA (ph = 8) TBE pufr (ph = Ř,γ): 0,ř ε Tris, 0,ř ε kyselina boritá, β mε Na 2 EDTA Pro p ípravu roztok byla pouţita deionizovaná voda (Direct-Q, Millipore, USA). 46
48 7.3. Přístrojové vybavení laboratoře a software Centrifuga Hettich Mikro β00r (Hettich, Velká Británie) Centrifuga Jouan BR4i (Thermo Fischer Scientific, USA) Centrifuga 5δ15 D (Eppendorf, Německo) Centrifuga PK110 (ALC, Italy) Genetický analyzátor ůbi PRISε γ100 (ůpplied Biosystems, USů) δaminární sterilní box ůura Bγ (Bioair Instruments, Itálie) εrazící box na -Ř0 C (Sanyo, USů), lednička s mrazícím boxem na -β0 C (δiebherr, Německo) Ph metr γ510 (Jenway, Velká Británie) Software i-control 1.6 (Tecan, Švýcarsko) Software winfast PVR2 (Leadtek Research Inc., Japonsko) Spektrofotometr Infinite 200 Nanoquant (Tecan, Švýcarsko) Systém pro p ípravu deionizované vody o čistotě vhodné pro tkáňové kultury (εillipore, USA) Termoblok Bio TBD-100 (Biosan, δotyšsko) Termocyklér Mastercycler 5330 (Eppendorf, Německo) Termocyklér εj εini Personal Thermal Cycler (Biorad, USA) Transluminator (Vilber Lourmat, Francie) UV transluminátor s dokumentačním systémem (Ultralum, USA) Vortex Bio V1 (Biosan, δotyšsko) Zdroj elektrického napětí Standard Power Pack Pβ5 (Biometra, Německo) 47
49 8. Pracovní postupy 8.1. Molekulárně genetická analýza Pro účely molekulární diagnostiky se nejčastěji pouţívá periferní krev v K 3 EDTA. Kyselina ethylendiamintetraoctvá vyvazuje vápenaté ionty, které jsou d leţité pro aktivaci koagulační kaskády. Nedoporučuje se pouţívat zkumavky s heparinem, který interferuje s Taq DNů polymerázou a m ţe tak negativně ovlivnit PCR amplifikaci Izolace DNA z krve (fenol-chloroformová extrakce) DNA vyizolujeme klasickou fenol-chloroformovou extrakcí z periferní krve. Rozpuštěnou DNů uchováváme do doby analýzy na -80 C ml EDTA krve doplníme do 50 ml vychlazeným lyzačním roztokem, promícháme 2. Dáme na γ0-δ0 min na led za občasného promíchání, dokud nedojde k lýzi červených krvinek 3. Centrifugujeme 10 min., γ000 rpm, δ C 4. Odsajeme supernatant a sediment rozsuspendujeme v 15 ml lyz. roztoku 5. Centrifugujeme 10 min., γ000 rpm, δ C 6. Odsajeme supernatant a sediment resuspendujeme v 15 ml PBS, ph = 7,4 7. Centrifugujeme 10 min., γ000 rpm, δ C 8. Odsajeme supernatant a sediment resuspendujeme v 4,5 ml STE pufru (ph 7,4) a p eneseme do 15 ml flakonky 9. P idáme β50 µl roztoku proteinázy K a β50 µl 10% SDS 10. Necháme p es noc ve vodní lázni na γ7 C 11. K natrávenému obsahu p idáme 5 ml zásaditého fenolu 12. Dáme 10 min. t epat, poté 10 min. na led 13. Centrifugujeme 10 min., γ500 rpm, δ C 14. K odebrané horní fázi p idáme stejný objem fenolu 15. Dáme 10 min. t epat, poté 10 min. na led 16. Centrifugujeme 10 min., γ500 rpm, δ C 17. K odebrané horní fázi p idáme stejný objem vychlazeného chloroformu 48
50 18. Dáme 10 min. t epat, poté 10 min. na led 19. Centrifugujeme 10 min., γ500 rpm, δ C 20. K odebrané horní fázi p idáme dvojnásobný objem ř6% vychlazeného ethanolu 21. Vzorky umístíme p es noc na -β0 C 22. Vysráţenou DNů zcentrifugujeme 15 min., γ500 rpm p i δ C 23. Pelet resuspendujeme v 1 ml 70% ethanolu 24. Centrifugujeme 5 min., rpm, δ C 25. Odstraníme supernatant, p idáme 1 ml 70% ethanolu a zvortexujeme 26. Centrifugujeme 5 min., rpm, δ C 27. Odstraníme supernatant a DNů necháme sušitp i laboratorní teplotě 28. DNů rozpustíme ve β00 µl 10 mε Tris, ph = 8,0 Takto p ipravenou DNů pouţijeme do PCR reakce nebo uchováváme p i -Ř0 C Příprava zásaditého fenolu pro izolaci: 1. Ve vodní lázni (50 C) necháme roztát krystalický fenol (δ0 ml) a p idáme hydroxychinolin (0,1 %, w/v) 2. Poté smícháme β0 ml fenolu s β0 ml 1ε Tris ph = 7,0-7,5 3. Prot epeme a necháme p es noc p i δ C 4. Centrifugujeme 5 min p i γ000 rpm 5. Odstraníme horní fázi a p idáme β0 ml β00 mε Tris, ph = 8,0-8,5 6. Prot epeme a necháme p es noc p i δ C 7. Centrifugujeme 5 min p i γ000 rpm 8. Odstraníme horní fázi a necháme stát 1 hod p i laboratorní teplotě 9. Centrifugujeme 5 min p i γ000 rpm 10. Nakonec p idáme 10 ml β00 mε Tris, ph = 8,0-8, PCR Vlastním principem polymerázové etězové reakce je in vitro namnoţení (amplifikace) specifického úseku nukleové kyseliny DNů. Praktické provedení reakce spočívá v cyklickém st ídání denaturace dvou etězcové nukleové kyseliny, p ipojení (annealing) specifických komplementárních oligonukleotid (primer ) a jejich prodlouţení (extenze) termostabilní polymerázou. Oligonukleotidové primery hybridizují s 49
51 protich dnými vlákny denaturované DNů, od jejichţ γ'-konc je zahájena syntéza komplementárních etězc. Syntéza nové DNů je katalyzována DNů polymerázou, v reakčním prost edí jsou p ítomny deoxynukleotidy, které p edstavují stavební kameny pro nově syntetizovanou DNA (Pr ša, 1řř7). Pro p ípravu byl pouţit HotStarTaq master mix od firmy Qiagen. Komerčně p ipravené primery byly navrţeny pomocí programu Primerγ Input (verze 0.δ.0) a Primer- BLAST. 1. P ipravíme si PCR reakční směs podle Tab. 2 (jako negativní kontrola pro proběhnutí PCR reakce slouţí vzorek bez DNů); sekvence jednotlivých primer je uvedena v Tab. 3 aţ Ř 2. Zvortexujeme a stočíme 3. Vzorky vloţíme do termocykléru a nastavíme odpovídající podmínky PCR reakce podle Tab. 9 nebo 10; po závěrečné fázi jsou vzorky ochlazeny na δ ºC 4. PCR produkty jsou analyzovány gelovou (agarózovou) elektroforézou Tab. 2: Sloţení PCR reakční směsi (1 vzorek) Komponenta Množství (µl) HST mix 12,5 H 2 O 9,5 Primer forward 1,0 Primer reverse 1,0 DNA 1,0* Pozn.: * 1 µl DNů u vzork s koncentrací niţší neţ δ00 ng/µl 0,5 µl u koncentrovanějších vzork Tab. 3: Sekvence pouţitých primer pro amplifikaci exon a hranic exon a intron genu HBB Název primeru Exon Sekvence 108_F 1,2 5 -GCCAAGGACAGGTACGGCTGTCATC-γ 109_R 1,2 5 -CCCTTCCTATGACATGAACTTAACCAT-γ HBBe3_F 3 5 -GGCTGGATTATTCTGAGTCCAAGC-γ HBBe3_R 3 5 -GCACTGACCTCCCACATTCC-γ 50
52 Tab. 4: Sekvence pouţitých primer pro amplifikaci exon genu CYB5R3 Název primeru Exon Sekvence DIA_2F 2 5 -GTGGGCACCAAAATGGATGA-γ DIA_2R 2 5 -CTGGGCAATGCTGTGATGCT-γ DIA_3F 3 5 -CAGCGTGCATGTGTGTGCAT-γ DIA_3R 3 5 -CCTGCTCTCCCTGGTGGAAA-γ DIA_4F 4 5 -AGCTGGGACGTGAGGGGAAT-γ DIA_4R 4 5 -GGGAGCACTGGGTCTTTGGA-γ DIA_5F 5 5 -ATTTGACGGCTGGGTGGATG-γ DIA_5R 5 5 -CAGGAGTGGCCTGTCTGCAA-γ DIA_6F 6 5 -TCCTCATGGCTCGCGTTAGAA-γ DIA_6R 6 5 -TAGCTCCCAGGCACTCAGAACC-γ DIA_7F 7 5 -GGTTCTGAGTGCCTGGGAGCTA-γ DIA_7R 7 5 -CCGCCAGTGCACAGAACATT-γ DIA_8F 8 5 -CCTCCTGAAAGCTCCGCAAG-γ DIA_8R 8 5 -TGTCGTCCAAAGGCTCAACG-γ DIA_9F 9 5 -GCTGGGATCAGCCTCTCCATT-γ DIA_9R 9 5 -ATCTGGGACACAGCCCTGCT-γ Tab. 5: Sekvence pouţitých primer pro amplifikaci exon genu HFE Název primeru Exon Sekvence HFE_1F 1 5 -TGTAGGGTGACTTCTGGAGCCAT-γ HFE_1R 1 5 -GCAAACTCCCAAGCGCAAAG-γ HFE_2F 2 5 -GGCCTGTTGCTCTGTCTCCA-γ HFE_2R 2 5 -AAAGCTCTGACAACCTCAGGAAGG-γ HFE_3F 3 5 -GGTCTTGTGGGAGCAGGGAA-γ HFE_3R 3 5 -CAGCCTCAGCCACCTCTGAAA-γ HFE_4F 4 5 -AAAGGGTATTTCCTTCCTCCAACC-γ HFE_4R 4 5 -GCAGATCCTCATCTCACTGCCA-γ HFE_5F 5 5 -GGGGTTGAGAGGAGTGCCTG-γ HFE_5R 5 5 -GCTGCTGGTGTCCCCAAAGA-γ HFE_6F 6 5 -TGGGTGAATGAGGAAAATAAGG-γ HFE_6R 6 5 -AGGTTCAACTCTCTCCTGAAACA-γ 51
53 Tab. 6: Sekvence pouţitých primer pro amplifikaci exon genu HFE2 (p vodně HJV) a HAMP Název primeru Exon Sekvence HJV_2F 2 5 -GGGCTTCCGGTCAAAATTCA-γ HJV_2R 2 5 -CTCACATGCCCACCCCTACA-γ HJV_3F 3 5 -GGAAGGCTGGAAACCCCTGA-γ HJV_3R 3 5 -AGTGGGGATGGGGAGGAATG-γ HJV_4aF 4 5 -GGAGAAGGGATCAAGGATTGAGG-γ HJV_4aR 4 5 -CCGACGATTGCGCTCTGAT-γ HJV_4bF 4 5 -CCCTGGGAACCATGTGGAGA-γ HJV_4bR 4 5 -TCTCCTAGGCCCTGCTTCCT-γ HAMP_1F 1 5 -AGGCCCCATAAAAGCGACTG-γ HAMP_1R 1 5 -TGCTCAGTGCTCGGGTGTCT-γ HAMP_2-3F 2,3 5 -CCACTTGGAGAGGAGCAGGT-γ HAMP_2-3R 2,3 5 -TCGGCAGAGAGAAAGGACAACA-γ Tab. 7: Sekvence pouţitých primer pro amplifikaci exon genu TFR2 Název primeru Exon Sekvence TFR2_1F 1 5 -TCCTAGGGCTCCCACAAACG-γ TFR2_1R 1 5 -TGGGAAGAAGCGAGGTCAGG-γ TFR2_2F 2 5 -CTGACCTCATTATTGCCAGATGC-γ TFR2_2R 2 5 -CCACCTCTGGACCCCAGAAA-γ TFR2_3F 3 5 -CCCAGAAGTGAAGGGCTTGG-γ TFR2_3R 3 5 -GCAGATGGGAGGACTCAGGA-γ TFR2_4F 4 5 -TCTGGCATCCTTCCCTCTT-γ TFR2_4R 4 5 -GACCCAGTGCAGGGTGTT-γ TFR2_5,6F 5,6 5 -GGCTGCAATTCCCGGAT-γ TFR2_5,6R 5,6 5 -TTCTCACTGGCAGTCCGACC-γ TFR2_7,8F 7,8 5 -TTGCCATTTCCTGGTTTCTC-γ TFR2_7,8R 7,8 5 -AGTTCACCCACAATCACCCT-γ TFR2_9F 9 5 -TGACTGCACTGCCCTACCCA-γ TFR2_9R 9 5 -CCCCTATCTTGCCAGGGTGT-γ TFR2_10F GTTGCCGGAATTGTGATGGG-γ 52
54 Tab. 8: Sekvence pouţitých primer pro amplifikaci exon gen SLC40A1 a FTL1 Název primeru Exon Sekvence SLC_1F 1 5 -TTGACGGGAGCTCGTCTCG-γ SLC_1R 1 5 -TGGTTCACAGCAGAGCCACA-γ SLC_2F 2 5 -TGTACGTGGTTTGTCCTGCAAA-γ SLC_2R 2 5 -TCATGGGGAAAGATCTTCGATG-γ SLC_3F 3 5 -GCCAGGAAGTGCCCTTTTGAT-γ SLC_3R 3 5 -CCCTCAAGTGTGGCATGCAG-γ SLC_4F 4 5 -ACTGGTCTGGTGTGATCCTCTGG-γ SLC_4R 4 5 -GAGTGCCTGTTGTGGGCAAA-γ SLC_5F 5 5 -CAGGTTAGGACATTATGCCCATTG-γ SLC_5R 5 5 -TCCATTTGCCAAGTTTGTGTAGGA-γ SLC_6F 6 5 -TGGGACTTGACCCAAACAACAA-γ SLC_6R 6 5 -CCCTAGATCCTCAAAAGCCTGCTC-γ SLC_7AF 7 5 -CCCATTGGGAAGGGGAATAGAA-γ SLC_7AR 7 5 -TCAAAAGGAGAAACGGACAAGTCC SLC_7BF 7 5 -TGGGAGCATCAGCTATAACTGGAAT-γ SLC_7BR 7 5 -GGGTAACAGAGCAAGACCCTGATC-γ SLC_8F 8 5 -TCCAGGCAAGACAGCTTTATTGTT-γ SLC_8R 8 5 -GAGCAAAACACCCAGCCATTT-γ FTL_1F 1 5 -CTCCTGCCACCGCAGATTG-3 FTL_1R 1 5 -TTTACCCGACCGCACAAAGAA-γ FTL_2F 2 5 -GCCCCTGGCCCTAATTTCC-γ FTL_2R 2 5 -CCGAACTCAATCTCCCAGAAGC-γ FTL_3F 3 5 -ACAAGCTGTCACATGTCTTTGTGG-γ FTL_3R 3 5 -GCAGCCAGTTGCAGATTAAAATGT-γ FTL_4F 4 5 -GCCTCATTTCACACCTGTCACA-γ FTL_4R 4 5 -CATCCCAACCCAGCCAGAAG-γ 53
55 Tab. 9: Časový a teplotní profil PCR reakce - amplifikace úsek genu HBB Teplota (ºC) Čas Počáteční denaturace min Denaturace s ůnnealing (nasednutí s primer ) Elongace (syntéza 72 1 min nového etězce) Extenze (konečná 72 7 min syntetická fáze γδ cykl amplifikace Tab. 10: Časový a teplotní profil PCR reakce - amplifikace úsek genu CYB5R3 a gen pro HH Teplota (ºC) Čas Počáteční denaturace min Denaturace s Annealing s Elongace s Extenze 72 7 min γβ cykl amplifikace PCR produkt je t eba p ed sekvenováním zbavit neinkorporovaných dntp a primer. P ítomnost primer by zp sobila nečitelnou směs sekvencí a dntp by změnily poměr koncentrace dntp/ddntp. 1. P ipravíme si 1% agarózový gel rozpuštěním agarózy v 0,5xTBE pufru a p idáním etidium bromidu (0,02 mg/ml) 2. Na gel naneseme vzorky: k β5 µl PCR vzorku p idáme 5 µl DNů loading pufru, jako marker pouţijeme β-log DNA ladder v mnoţství 10 µl 3. Necháme proběhnout elektroforézu p i napětí (1-10 V/cm gelu) po dobu 1-2 hodiny 54
56 4. DNů produkty detekujeme prost ednictvím UV transluminátoru s dokumentačním systémem a softwarem WinFast PVRβ 5. Následně produkty vyizolujeme z gelu pomocí komerčního kitu εini Elute Gel Extraction Kit (Qiagen, Německo, support/resource-center/resource-download.aspx?id=fa2ed17d-a5e c1-3d0ea6c2287d&lang=en) Sekvenování DNA V dnešní době se pro vysokokapacitní sekvenace vyuţívá výlučně metoda publikovaná Sangerem v roce 1ř75. Sekvenační reakce je zaloţena na inkorporaci terminačních fluorescenčně značených dideoxynukleotid (ddntp). Kaţdý z ddntp má navázaný jiný fluorochrom, který je moţné odlišit na základě r zných emisních spekter. Za azení ddntp do etězce zp sobí ukončení syntézy molekuly DNů. V reakci vzniká směs r zně dlouhých produkt. Po p ečištění od volných nukleotid je vzorek rozdělen na kapilární elektroforéze, která je součástí genetického analyzátoru. Fotooptický systém p ístroje zajistí automatické p ečtení sekvence, se kterou je moţné díky p íslušnému softwaru dále pracovat. ůutomatické stanovení sekvence nukleotid DNů metodou kapilární elektroforézy s laserovým detektorem. Z jednoho stanovení je obvykle čitelných bází. Výkonnost p ístroje, který byl pouţit (genetický analyzátor ůbi PRISε 3100), je aţ bází/den. A. Sekvenační reakce 1. P ipravíme si sekvenační reakční směs podle Tab. 11; sekvence jednotlivých primer je uvedena v Tab. γ aţ Ř 2. P idáme vypočítané mnoţství templátu (produkt vyizolovaný z gelu) na základě koncentrace a velikosti PCR produktu, Tab Vzorky zvortexujeme a stočíme 4. Následně vloţíme do termocykléru a nastavíme odpovídající podmínky pro sekvenační reakci podle Tab
57 Tab. 11: Sloţení reakční směsi pro cyklické sekvenování (1 vzorek) Komponenta Množství Templát (PCR produkt) 1-30 ng Master Mix [2,5X] 1,0 µl Sekvenční pufr [5X] 1,5 µl Primer F/R [10 µm] 0,5 µl TK H 2 O do 10 µl Pozn.: mnoţství templátu dle Tab. 12 F/R: forward/reverse Tab. 12: Koncentrace produktu pro sekvenační reakci dle velikosti PCR produktu PCR produkt [bp] Koncentrace produktu [ng] Tab. 13: Časový a teplotní profil sekvenační reakce Teplota (ºC) Čas Počáteční denaturace 96 1 min Denaturace s Annealing 50 5 s Elongace 60 4 min Extenze min βδ cykl B. Přečištění sekvenační reakce 1. Sekvenační produkty dále p ečistíme komerčním kitem Big Dye X Terminator Purification kit (Applied Biosystems, UK): k sekvenačnímu produktu p idáme δ5 µl Sůε roztoku a 10 µl zvortexovaného XTerminatoru 2. Vortexujeme 30 min. 3. Centrifugujeme 2 min., 1000 rpm, RT µl odebraného supernatantu p eneseme na sekvenační destičku do doby analýzy uchováváme v temnu p i δ C 56
58 C. Sekvenační analýza Pro sekvenační analýzu byl pouţit genetický sekvenátor ůbi PRISε γ100 ( P ístroj pracuje na principu kapilární elektroforézy. Obsahuje 16 kapilár o délce γ6 cm naplněných polymerem POP-4. Vzorek je unášen elektroforetickým pufrem (10X Genetic ůnalyzer buffer) s EDTA. ůnalýza probíhá p i teplotě 55 C. Kapilára a kladná elektroda je pono ena do jamky se vzorkem, 10 µl je injektováno do kapiláry. Díky vloţenému napětí na elektrodě (1β,β kv), záporně nabité fragmenty DNů postupují kapilárou směrem k anodě. Kapilára je oza ována ze β stran argonovým laserem (excitační vlnová délka δřř-415,5 nm). Fragmenty DNů obsahující fluorescenční značku emitují pohlcené zá ení. Spojité spektrum je snímáno CCD kamerou (chargecoupled device). Software vyhodnotí emisní spektra a p evádí je do podoby barevných k ivek (elektroferogram) (Pospíšilová, β01β). Výsledné elektroferogramy vyhodnotíme pomocí programu FinchTV nebo Chromas a genomové databáze ENSEεBδ a vyhledávači BδůST ( Enzymová analýza Pro enzymové analýzy byla pouţita periferní krev s heparinem sodným Příprava lyzátu pro enzymovou analýzu P ipravíme si jednoduchou kolonku skládající se z graduovaného válce injekční st íkačky, filtračního papíru a směsi celulosy. Kolonka nám zajistí odseparování leukocyt a trombocyt. Pracujeme na ledě. Příprava kolonky: 1. Smícháme ekvivalentní váhové mnoţství mikrokrystalické celulosy a α-celulosy s 0,154 M roztokem NaCl (dále jen roztok NaCl) 2. Z filtračního papíru vyst ihneme kolečko a vloţíme ho na dno graduovaného válce injekční st íkačky 3. Navlhčíme jej roztokem NaCl pro lepší p ilnutí k povrchu 4. Poté válec vloţíme do 15 ml zkumavky a naplníme jej suspenzí z celulosy do 57
59 výšky cca β cm Příprava hemolyzátu: 1. 1 ml periferní krve naneseme na kolonku 2. Necháme volně eluovat kolonkou vychlazeným fyziologickým roztokem 3. Vzniklý efluent na edíme vychlazeným fyziologickým roztokem do konečného objemu cca 8 ml 4. Centrifugujeme 5 min p i β500 rpm a δ C 5. Odsajeme supernatant 6. Pelet na edíme 1:1 vychlazeným fyziologickým roztokem a následně k němu p idáme stabilizační roztok v poměru 1:ř 7. Takto p ipravený hemolyzát pouţijeme buď p ímo pro stanovení aktivity cytochrom-b5-reduktázy nebo zamrazíme a uchováváme v alikvotech na -80 C (Beutler, 1984) Spektrofotometrické stanovení aktivity cytochrom-b5-reduktázy 1. P ipravíme si reakční směs: 0,2 mm NADH v 0,1 M Tris-HCl/0,5 mm EDTA (ph 8,0), a inkubujeme 10 min. p i γ0 C 2. Zároveň si p ipravíme směs lyzátu (1:20) s roztokem 2 mm K 3 Fe(CN) 6 (1:10 lyzátu) a necháme proběhnout oxidaci hemoglobinu v roztoku po dobu inkubace reakční směsi 3. K reakční směsi p idáme směsný roztok lyzátu s ferrikyanidem (1/10) 4. P evedeme β00 µl na 96-jamkovou mikrotitrační destičku pro UV 5. Mě íme absorbanci p i vlnové délce γδ0 nm v 1minutových intervalech po dobu 20-γ0 min. a teplotě γ0 C 6. Výsledkem je k ivka závislosti absorbance na čase, z rozdíl změny absorbance po odečtení kontroly vypočítáme pomocí δambert-beerova zákona specifickou aktivitu daného enzymu: 58
60 a (µmol.min -1 ) = ( A x V/( x l x t)) x f, kde: ů: změna absorbance v čase (odečtení kontroly) V: objem reakční směsi 200 µl; l: délka mě ené vrstvy (0,58 cm) : absorbanční koeficient NůD(P)H p i γδ0 nm (6ββ0 l.mol -1.cm -1 ) t: čas (min) f: faktor edění. Specifická aktivita, sa (U/g Hb) = ( A x 0,028 x f )/ m Hb, mhb: mnoţství hemoglobinu ve stanovovaném vzorku lyzátu, U = 16,67 nkat (µmol.min -1 ) Stanovení koncentrace hemoglobinu v enzymovém lyzátu Koncentraci hemoglobinu stanovujeme spektrofotometricky. Hemoglobin má absorbční spektrum v oblasti viditelného světla, absorpční maximum má p i δ1δ nm. Vše mě íme v trojím opakování. 1. Hemolyzát na edíme δ0-ř0krát TK H 2 O 2. β00 µl vzorku p eneseme na ř6-jamkovou mikrotitrační destičku (3x200 µl) 3. Mě íme absorbanci p i δ1δ nm 4. Vypočteme celkovou koncentraci Hb dle: C g/l = ( A/l x ) x Mr x f, kde: ů: změna absorbance (vzorek minus blank (voda)) l: délka mě ené vrstvy (0,58 cm) : absorbanční koeficient Hb p i δ1δ nm (5βδβŘ0 l.mol -1.cm -1 ) εr: molekulová hmotnost Hb (64500 g.mol -1 ) f: faktor edění (Prahl, 2008). 59
61 9. Výsledky 9.1. talasemie Soubor pacient Soubor pacient s podez ením na suspektní talasemii obsahoval 1Řγ pacient (100 ţen a Řγ muţ ) ze 10δ nep íbuzných rodin pocházejících z r zných míst České a Slovenské republiky. V době diagnózy bylo v souboru γř dětí do 1Ř let (17 dívek a 21 chlapc ). V p ípadě potvrzení diagnózy na molekulární úrovni identifikací kauzální mutace v genu HBB byli vyšet eni i další členové rodiny. Dosaţené výsledky talasemie minor bývá někdy doprovázena nápadnou kompenzační erytrocytózou s počtem erytrocyt nad 6,0x10 12 /l (Divoký, β005). Toto zjištění potvrzují i mnou získané údaje. Z celkového počtu 1Řγ pacient mělo hodnoty erytrocyt v krevním obraze nad 6,0x10 12 /l celkem δ0 pacient tj. cca ββ %. Podrobněji tyto výsledky shrnuje Tab. 14 a Tab. 15. Základní parametry krevního obrazu byly vyšet eny na HOK FN Olomouc s pouţitím automatického analyzátoru (Sysmex XE-500, Sysmex, Kobe, Japan). Tab. 14: Počet erytrocyt u muţ. Počet erytrocytů (x10 12 /l) Počet mužů Poměrné zastoupení jedinců (%) < 4,00 1 1,20 4,00 4,50 3 3,61 4,51 5,00 2 2,41 5,01 5, ,48 5,51 6, ,96 6,01 6, ,87 6,51 7, ,05 > 7, ,41 celkem Pozn.: Referenční hodnoty pro počet erytrocyt (muţi): δ,00-5,80x10 12 /l ( 60
62 Tab. 15: Počet erytrocyt u ţen. Počet erytrocytů (x10 12 /l) Počet žen Poměrné zastoupení jedinců (%) < 4, ,00 4, ,51 5, ,01 5, ,51 6, ,01 6, ,51 7, > 7, celkem Pozn.: Referenční hodnoty pro počet erytrocyt (ţeny): 3,80-5,20x10 12 /l ( Pro talasemie je rovněţ charakteristický projev anémie, kdy sníţení koncentrace hemoglobinu pod normu stanovenou dle věku a pohlaví je výsledkem sníţené tvorby jednoho nebo více polypeptidových etězc globinu. Na základně hodnot hemoglobinu je pak moţné míru anémie v rámci našeho souboru pacient rozdělit dále na lehkou nebo st edně těţkou formu (Fábryová, β007) většina pacient, muţského i ţenského pohlaví (81 resp. 73 %), má projevy mírné anémie tj. hodnoty Hb nad ř6 g/l (Tab. 16 a Tab. 17). Tab. 16: Hodnoty hemoglobinu u muţ. Koncentrace hemoglobinu v krvi (g/l) Počet mužů Zastoupení jedinců (%) norma: ,05 lehká anémie: ,72 středně těžká anémie: < ,23 cekem
63 Tab. 17: Hodnoty hemoglobinu u ţen. Koncentrace hemoglobinu v krvi (g/l) Počet žen Zastoupení jedinců (%) norma: lehká anémie středně těžká anémie: < cekem P i diferenciální diagnostice jsou dalšími faktory, na které se p ihlíţí, mnoţství HbA 2, který bývá zvýšený u talasemie minor (nad γ %), a HbF, zvýšený zejména u talasemií, ale i v některých p ípadech u talasemií ( 1,1 %). Ve všech stanovovaných p ípadech (tj. v počtu167 vyšet ení) byly hodnoty HbA 2 zvýšeny, a to v rozmezí γ,0-7,4 %; v cca 50 % jeho hodnoty dosahovaly 3 5 % (Tab. 18). Hodnoty HbF a HbA 2 byly rovněţ stanoveny na HOK FN Olomouc (HbA 2 kvantifikováno pomocí kolonek od firmy Helena Biosciences a p i stanovení HbF se vyuţívá jeho zvýšené rezistence k alkalické denaturaci ve srovnání s ostatními hemoglobiny). Tab. 18: Hodnoty HbA 2 u muţ i ţen. Hodnota HbA 2 (%) Počet Zastoupení jedinců % 3,1 5, ,55 5, ,70 neměřeno 16 8,74 celkem Hodnota HbF byla stanovena jen u řř pacient. U zbylých pacient byla tato potenciálně pomocná diagnostická informace povaţována za nadbytečnou, neboť byla v dané rodině jiţ d íve potvrzena nějaká talasemická mutace sekvenováním. Namě ené 62
64 hodnoty HbF se pohybovaly v rozmezí 0,1 1ř %; zvýšená hodnota HbF ( 1,1 %) se vyskytovala ve δδ p ípadech z celkových řř mě ených (Tab. 19). Tab. 19: Hodnoty hemoglobinu F v souboru pacient. HbF (%) Počet Zastoupení jedinců % normál 1 % 40 21,86 1,0 3,0 % 34 18,58 3,1 5,0 % 6 3,28 5 % 4 2,19 neměřeno 99 54,10 celkem První molekulárně-genetickou charakteristiku talasemií u rodin českého a slovenského p vodu publikoval prof. Indrák se svojí skupinou jiţ v roce Na tuto studii navázalo několik dalších; poslední z nich byla publikována v roce β005 (Divoký et al., 2005). Náš soubor susp. talasemických pacient vznikl v letech Za posledních Ř let bylo retrospektivně molekulárně-genetickými metodami vyšet eno 1Řγ osob ze 10δ nep íbuzných rodin, identifikovali jsme celkem 15 r zných exonových nebo intronových mutací globinového genu (HBB) v heterozygotním stavu, jednu krátkou deleci δδ nukleotid a t i polymorfismy, dva časté, CD2 (C-T) (rs713040) a IVS-II-666 (rs ) a jeden vzácný, +96 c.*+96t > C (3 ' UTR+1570 T > C) (rs ). Výskyt homozygot v naší populaci je málo pravděpodobný, i kdyţ není zcela vyloučený. Nejčastěji se vyskytovaly mutace mediteránního p vodu. Výsledky zahrnující talasemické mutace detekované v rámci této studie jsou shrnuty v Tab. 20. Jak vyplývá z tabulky, nejčetněji se v rámci našeho souboru 1Řγ pacient vykytují mutace IVS-I-110 (G-A), CD 39 (C-T), IVS-I-1(G-A) a IVS-I-6 (T-C); p edstavují cca 67 % všech talasemických mutací detekovaných v této studii. Jedná se zároveň o jedny z nejčastějších mediteránních mutací (Indrák, 1993). Ostatní mutace jsou pak zastoupeny v menší mí e. Nejfrekventovanější mutace IVS-I-110 (G-A), záměna guaninu za adenin, se nachází β1 nukleotid p ed konvenční nukleotidovou sekvencí sest ihu (5 donorovým místem 63
65 sest ihu) (Obr. 8). Tato intronová mutace zp sobuje vznik nového sest ihového místa (AG^GC), které pak následně vede z 80 % k abnormálnímu sest ihu mrnů a jen z 20 % k normálnímu sest ihu mrnů (Indrák, 1řřγ). Zp sobuje + talasemii. Tab. 20: talasemické mutace detekované u vyšet ovaného souboru 183 pacient ze 105 rodin z české a slovenské populace. Mutace HGVS nomenklatura Počet osob (rodin) Frekvence % Fenotyp Původ IVS-I-110 (G-A) c.93-21g>a 42 (26) 22,95 + med. CD 39 (C-T) c.118c>t 33 (14) 18,03 0 med. IVS-I-1 (G-A) c.92+1g>a 26 (17) 14,21 0 med. IVS-I-6 (T-C) c.92+6t>c 22 (12) 12,02 + med. IVS-II-1 (G-A) c.315+1g>a 11 (5) 6,01 0 med. CD 8 (-AA) c.25_26delaa 10 (5) 5,46 0 med. CD 38/39 (-C) c.118delc 10 (7) 5,46 0 český IVS-II-745 (C-G) c c>g 8 (5) 4,37 + med. CD 121 (G-T) c.364g>t 4 (3) 2,19 0 slovenský/český CD 52 (-A) c.158dela 3 (1) 1,64 0 český CD 5 (-CT) c.17_18delct 2 (1) 1,09 0 med. CD 7/8 (+G) c.24_24dupg 2 (1) 1,09 0 slovenský CD 17 (A-T) c.52a>t 2 (2) 1,09 0 asijský CD 45 (-T) c.136delt 2 (1) 1,09 0 pákistánský -44 nt delece c.76_92+27del 2 (1) 1,09 0 ecký CD 41/42 (- TTCT) c.126_129delcttt 1 (1) 0,55 0 asijský Pozn.: HGVS: human genom variation society med.: mediteránní p vod (HBB accession: NM_ ) 64
, je druhou nejčastější talalasemickou mutací v našem souboru (Tab. 20). Zp sobuje 0 talasemii. Tuto mutaci adíme mezi nesmyslné mutace.")
. Obr. 9: Mutace CD39 (C-T), v 2 exonu HBB genu. Druhá nejčastější mutace v této studii (γγ pacient /14 rodin).")
66 Obr. 8: Mutace IVS-I-110 (G-ů). Nejčastější mutace v této studii detekována u δβ pacient z β6 rodin. Mutace CD 39 (C-T), záměna cytosinu za thymin, v kodonu γř ve druhém exonu -globinového genu (Obr. 9), je druhou nejčastější talalasemickou mutací v našem souboru (Tab. 20). Zp sobuje 0 talasemii. Tuto mutaci adíme mezi nesmyslné mutace. Ovlivňuje translaci RNů a vede k p edčasnému za azení STOP kodonu do kódující sekvence a tím k p edčasné terminaci globinového etězce. Vzniklý etězec je o 107 aminokyselin kratší neţ jeho normální varianta (146 aminokyselin). Obr. 9: Mutace CD39 (C-T), v 2 exonu HBB genu. Druhá nejčastější mutace v této studii (γγ pacient /14 rodin). T etí nejběţnější talalasemická mutace v našem souboru je mutace IVS-I-1 (G-A), záměna adeninu za guanin, v akceptorovém místě sest ihu 1. intronu globinového genu (Obr. 10). Tato mutace zasahuje konvenční nukleotidovou sekvenci sest ihu, výsledkem je absence funkční mrnů. Výsledná 0 talasemická alela neprodukuje ţádný funkční globin. 65
.")
. Čtvrtá nejčastější mutace (ββ pacient /12 rodin).")
. Mutace IVS-II-1 (G-A) detekovaná u 11 pacient z 5 rodin zasahuje konvenční nukleotidovou sekvenci sest ihu,")
67 Obr. 10: Mutace IVS-I-1 (G-A). T etí nejčastější mutace v našem souboru (β6 pacient /17 rodin). Další detekovanou mutací je mutace IVS-I-6 (T-C). Tato intronová mutace sniţuje účinnost správného sest ihu mrna, zp sobuje + talasemii (Obr. 11). Obr. 11: Mutace IVS-I-6 (T-C). Čtvrtá nejčastější mutace (ββ pacient /12 rodin). εezi méně časté, ale detekované u více neţ 5 % pacient ze souboru pat í mutace IVS-II-1 (G-A), CD 8 (-AA) a CD 38/39 (-C). Mutace IVS-II-1 (G-A) detekovaná u 11 pacient z 5 rodin zasahuje konvenční nukleotidovou sekvenci sest ihu, brání tedy normálnímu sest ihu globinové mrnů. Výsledkem je absence funkční mrnů a vznik 0 talasemie (Obr. 12). Obr. 12: Mutace IVS-II-1 (G-A). 66
 a k ukončení translace za azením STOP kodonu v kodonu 21 (TGA). Vzniklý globinový etězec je o 125 aminokyselin kratší neţ normální.")
 (Obr. 14) vede ke vzniku terminačního kodonu na pozici 60 (vzniklý globinový etězec má jen 86 aminokyselin).")
68 U deseti pacient z pěti rodin byla popsána posunová mutace v CD 8 (-AA), exonu 1 HBB genu (Obr. 13). Tato mutace (delece) se často vyskytuje v Turecku a ůzerbajdţánu. Vlivem této delece dochází k posunu čtecího rámce ( frameshiftu ) a k ukončení translace za azením STOP kodonu v kodonu 21 (TGA). Vzniklý globinový etězec je o 125 aminokyselin kratší neţ normální. Obr. 13: Posunová mutace v CD 8 (-AA) v 1. exonu HBB genu. U 10 osob ze 7 českých rodin byla nalezena 0 talasemická mutace zp sobující deleci jednoho cytosinu v sekvenci ACC CAG v kodonech γř a γř. Tato posunová mutace 38/39 (-C) (Obr. 14) vede ke vzniku terminačního kodonu na pozici 60 (vzniklý globinový etězec má jen 86 aminokyselin). Tato mutace byla poprvé v České republice popsaná Indrákem et al. (1993b). Obr. 14: Posunová mutace CD 38/39 (-C) v 3. exonu HBB genu. U t í rodin byla potvrzena vzácná mutace v CD 121 (G-T) v exonu 3 HBB genu, která má v našich populacích nezávislý, česko/slovenský p vod (Indrák et al., 1992). Tato mutace vytvá í p edčasný terminační kodon, dochází ke zkrácení etězce, který tvo í pouze 1β0 animokyselin. Tato mutace je vzácná, její fenotypové projevy se liší od projev mutací v 1. a 2. exonu a v obou intronech. Nosiči s touto mutací mívají st edně těţkou aţ těţkou anémii a obraz talasemie intermedia. 67
, v 2 exonu genu HBB; nová mutace v české populaci.")
 v exonu 2 genu HBB.")
69 U t í osob z jedné rodiny byla prokázána v české populaci zatím nepopsaná mutace CD 52 (-A) v exonu 2 genu HBB (Obr. 15). Všichni pacienti mají zvýšené hodnoty HbA 2 a to v rozmezí 4,5-5,3 %. Jedná se o heterozygoty a hodnoty krevního obrazu odpovídají heterozygotnímu nosičství 0 talasemické alely. Obr. 15: Mutace CD 52 (-A), v 2 exonu genu HBB; nová mutace v české populaci. V jednom p ípadě byla identifikována talasemická mutace, která rovněţ nebyla v České republice doposud popsaná. Jedná se o deleci čty nukleotid (-CTTT) v kodonu 41,42 CD 41/42 (-TTCT) (Obr. 16). Tato mutace je asijského p vodu a byla nalezena u imigranta z Vietnamu. Obr. 16: Mutace CD 41/42 (-TTCT) v exonu 2 genu HBB. Jedná se o nově diagnostikovanou mutaci v České republice u pacienta vietnamského p vodu. U jednoho dětského pacienta s mikrocytární hypochromní anémií (RBC δ,β7 x /l, Hb 115 g/l, Hct γγ %, εcv 77,5 fl, εch β6,ř pg) a lehce zvýšenou hodnotou Hbů 2 (3,3 %), ale normální hodnotou HbF (β,7 %), byly prokázány γ polymorfismy: CD2 (C-T) (rs713040), IVS-II-666 (rs ), +96 c.*+96t > C (3 ' UTR+1570 T > C) (rs ). V p ípadě polymorfism rs a rs (Obr. 17a a 17b) se jedná o relativně časté polymorfismy. SNP rs je poměrně vzácný (Obr. 17c). Nachází se 68
 B) C) Obr. 17: T i polymorfismy u jednoho pacienta s talasemickým fenotypem. A) CD2 (C-T), B) IVS-II-666 a C) +96 c.*+96t v HBB genu. 9.2. Methemoglobinémie Soubor pacient Soubor pacient obsahoval γ pacienty s mírnou resp.")
70 v nep episované γ -UTR oblasti globinového genu (Divoký et al., 1994, Bilgen et al., 2011). Souvislost mezi talasemickým fenotypem a tímto vzácným SNiPem zatím nebyla jednoznačně potvrzena. A) B) C) Obr. 17: T i polymorfismy u jednoho pacienta s talasemickým fenotypem. A) CD2 (C-T), B) IVS-II-666 a C) +96 c.*+96t v HBB genu Methemoglobinémie Soubor pacient Soubor pacient obsahoval γ pacienty s mírnou resp. asymptomatickou methemoglobinémii (MetHb 7-11 %) (Tab. 21). V prvém p ípadě se jednalo o 15-ti letého chlapce s εethb 7 % (krevní obraz v normě). Viditelným projevem bylo namodralé zbarvení k ţe; účinky εethb jsou redukovány kys. askorbovou. εatka má MetHb v normě (0,ř %), otec není k dispozici. V druhém p ípadě šlo o náhodný záchyt 75leté ţeny s εethb 11 % bez klinických projev. T etím p ípadem bylo měsíční dítě narozené bez ikteru, ale opakovaně cyanotické se zvýšenou hladinou MetHb (β0 %). Po zahájení terapie kys. askorbovou došlo k poklesu MetHb na 8-11 %. Oba rodiče jsou asymptomatičtí s MetHb v normě. Ve všech p ípadech byla vyloučena hemoglobinopatie, deficit pyruvátkinázy i glukóza-6-fosfátdehydrogenázy (G6PD). Tab. 21: Pacienti se sníţenou aktivitou Cb5R biochemická a molekulárně-genetická charakterizace. Pacient 1 Pacient 2 Pacient 3 Věk/pohlaví 15/M 75/Ţ 1m/Ţ MetHb (%) Cb5R (U/g Hb) 3,0 n.d. 2,44 69
71 Mutace c.400a>g c.890g>a c.316g>a Exon AK záměna Met134Val Arg297His Val106Met Pozn.: m: věk v měsících; ε/ţ: muţ/ţena; n.d.: nestanovováno; ref. rozmezí pro aktivitu Cb5R: Ř,δβ-13,66 U/g Hb); gen CYB5R3 (22g13.2, NM_ ). Dosaţené výsledky U prvního pacienta byla stanovena sníţená aktivita Cb5R (30 %). εolekulárněgenetická analýza potvrdila, ţe je heterozygotem pro známý polymorfizmus (c.132g>a resp. p.pro44pro) a novou mutaci (c.400a>g resp. p.met134val) v 5. exonu genu CYB5R3 (Tab. 21, Obr. 18A, B). ůsymptomatická matka má normální aktivitu Cb5R a není nositelkou ani jedné ze substitucí. U druhé pacientky bylo prokázáno, ţe je heterozygotem pro substituci c.890g>a tj. p.arg297his v 9. exonu (Tab. 21, Obr. 18C). Stanovení aktivity Cb5R nebylo moţné zajistit. T etí pacientka má cca β5% aktivitu Cb5R a je heterozygotem pro mutaci c.316g>a (p.val106met) ve 4. exonu (Tab. 21, Obr. 18D). Oba rodiče mají rovněţ sníţenou aktivitu Cb5R (γ,δ a δ,ř U/g Hb, otec resp. matka). Otec je nositelem stejné mutace jako dcera, u matky i p es sníţenou aktivitu Cb5R nebyla mutace potvrzena. ůktivita Cb5R je výrazně ovlivněná i věkem pacienta; i u zdravých novorozenc a kojenc cca do γ měsíc ţivota je aktivita Cb5R cca 60 %. A) B) C) 70
, který je krátkou smyčkou propojen s NADH-vazebnou doménou.")

72 D) Obr. 18: Mutace v genu CYB5R3. A) SNP c.132g A (pacient 1) B) c.400g>a, exon 5 (pacient 1) C) c.890g>a, exon 9 (pacient 2) a D) c.316g A, exon 4 (pacient 3). Mutace Met134Val se nachází na C-konci beta- etězce FůD-vazebné domény (Obr. 19), který je krátkou smyčkou propojen s NADH-vazebnou doménou. Záměna by mohla vést k oslabení kontaktu mezi oběma doménami a ovlivnit tak stabilitu enzymu (Percy & Lappin, 2008). Mutace Met134Val nebyla dosud popsána. Substituce c.890g>a pro záměnu Arg297His je uváděna v databázi ( jako vzácný polymorfismus; je situována ve FůD-vazebné doméně v blízkosti vazebného místa pro NůDH a mohla by nep ímo ovlivňovat interakci mezi oběma kofaktory. Mutace c.316g>a (p.val106met) je stejně jako v p ípadě mutace Met134Val součástí FůD-vazebné domény (Obr. 19). Obr. 19: ůminokyselinová sekvence lidské erytrocytární NůDH-Cb5R s lokalizací detekovaného polymorfismu a aminokyselinových záměn. FůD a NůD(P)H-vazebná doména jsou vyznačeny zeleně a oranţově (Percy & Lappin, 2008). 71
73 9.3. Hemochromatóza Soubor pacient Soubor obsahoval 11 pacient s hemolytickou anémií v d sledku deficitu pyruvátkinázy prokázáném jak na biochemické tak molekulární rovni. Jeden pacient se vyznačoval zvášť těţkým fenotypovým projevem a to uţ od narození těţká forma hemolytické anémie (Hb 77 g/l) s hepatosplenomegalií a výskytem novorozenecké ţloutenky a s výraznou hyperferitinémií (feritin cca δ000 µg/l) a saturací transferinového receptoru dosahující prakticky 100 % (Tab. 22). I u ostatních pacient je patrná st ední aţ těţká forma hemolytické anémie (Hb g/l) s bilirubinémií a retikulocytózou. Ve většině p ípad byla potvrzena potrzena novorozenecká ţloutenka, aţ na jeden p ípad všichni podstupují nebo podstoupili výměnné transfuze. U 8 z nich byly patrné známky p etíţení ţelezem (Tab. 22). Tab. 22: Klinická data pacient s deficitem pyruvátkinázy. Pacient Pohlaví/věk Hb (g/l) Bilirubin (µmol/l) koincidence deficitu pyruvátkinázy s vrozenou poruchou metabolismu ţeleza. Výskyt 72 Ret (%) Feritin (µg/l) 1 M/ M/3m , F/ , M/10m ,5 303 n.d. 5 M/ , F/ , F/ , F/ , M/3m ,1 802 n.d. 10 F/ ,3 29, M/ , Pozn.: m: věk v měsících Hb: hemoglobin Ret: retikulocyty TfS: saturace transferinu referenční rozmezí pro bilirubin: 0-βγ µmol/l, Ret: 0,5-γ %, feritin: ţeny β00, muţi γ00 a děti 70 µg/l, TfS: β1-48 %. Dosaţené výsledky U pacienta s p etrávající hyperferitinémie byla provedena sekvenční analýza gen, jejichţ mutace zp sobuje vrozenou hemochromatózu, aby se vyloučila p ípadná TfS (%)
 ani TFR2 (transferinový receptor β).")


74 kauzální mutace pro hemochromatózu nebyl prokázán ani v jednom testovaném genu HFE, HFE2 (hemojuvelin), HAMP (hepcidin), SLC40A1 (feroportin 1) ani TFR2 (transferinový receptor β). Rovněţ u něj nebyla nalezena mutace v IRE oblasti genu pro L- feritin (FTL), která by zp sobovala vzácnou vrozenou poruchu metabolismu ţeleza hyperferitinemii s autozomálně dominatntní kongenitální kataraktou (HHCS, hereditary hyperferritinemia cataracta syndrome). U HHCS je katarakta výsledkem nadměrného hromadění feritinu, který je nekontrolovatelně translatován vlivem mutace v IRE oblasti, v oční čočce. Bylo u něj detekováno pouze 5 jiţ d íve popsaných SNPs a to v genu TFR2 (p.arg752his), SLC40A1 (c.44-24g C a c.-330cgg 8 v IVS a 5 -nep ekládané oblasti, p.val221val) a FTL (p.leu55leu) (Obr. 20). A) B) C) 73
75 Obr. 20: Pacient s p etrávající hyperferitinémií. Detekce popsaných SNPs: ů) heterozygotní mutace p.arg752his v genu TFR2 B) homozygotní mutace p.valββ1val v genu SLC40A, a C) homozygotní mutace p.leu55leu v genu FTL. U ostatních pacient ze souboru byl analyzován pouze HFE gen. Mutace p.his63asp byla potvzena u 4 z nich, ve γ p ípadech v heterozygotní formě, v 1 p ípadě v homozygotní formě a v 1 p ípadě v kombinaci s mutací p.cysβřβtyr (Tab. 23). Tab. 23: Mutace v genu PKLR a HFE u pacient s deficitem pyruvátkinázy. Pacient Mutace genu AK záměna PKLR HFE 1 Arg116_L117delinsGlnHisCys wt/wt /Arg116_L117delinsGlnHisCys 2 Arg518fs/Arg518fs wt/wt 3 Arg510Gln/Arg532Trp His63Asp/Cys282Tyr 4 Arg510Gln/Arg532Trp His63Asp/wt 5 Arg498His/Arg510Gln His63Asp/wt 6 Arg486Trp/Arg532Trp His63Asp/His63Asp 7 Arg532Trp/Arg532Trp wt/wt 8 Asp293Val/Arg510Gln wt/wt 9 Gly275Arg/Arg532Trp wt/wt 10 Arg510Gln/Arg510Gln wt/wt 11 Gly332Ser/Arg486Trp wt/wt Pozn.: pacient 3 a 4 jsou sourozenci wt: wild type alela. 74
76 10. Diskuze Beta talasemie jsou vrozené mikrocytární anémie, které vznikají v d sledku sníţené produkce nebo úplné absence -globinového etězce. Nejčastější p íčinou jsou bodové mutace v sekvenci globinového genu nebo jeho promotoru. εutantní globinové alely jsou ve st ední Evropě relativně vzácné, p esto jsou v České republice nejčastější p íčinou vrozené mikrocytární anémie (kap. 3.2). V České republice se problematikou talasemiií dlouhodobě zabývají hlavně vědečtí pracovníci na Hemato-onkologické klinice ve Fakultní nemocnici v Olomouci a na ÚHKT v Praze. Největší p ínos mají práce Indráka a Divokého, jejichţ výsledky dovedly diagnostiku talasemií v České republice na světovou úroveň, dále pak d ívější práce Chrobáka (FN Hradec Králové) a Brabce (ÚHKT Praha). Na Slovensku se této problematice začal věnovat Hrubiško (FNsP Bratislava), později ho následovala Sakalová a Fábryová (FNsP Bratislava). Jak uvádí ve své práci Indrák (1993), p i diagnostice talasemie jsou jedním z rozhodujících faktor některé parametry krevního obrazu zejména hodnoty počtu erytrocyt > 5,0 x /l, mikrocytóza MCV < 80 fl a hypochromie εch < β7 pg, zvláště jsou-li tyto změny p ítomny familiárně. Také nález zvýšené hodnoty Hbů 2 svědčí pro talasemii minor. Proto se také jeho stanovení vyuţívá p i diferenciální diagnostice talasemií. Tomu odpovídá i základní charakteristika našeho souboru nemocných se suspektní talasemií - 84 % pacient mělo zvýšené hodnoty erytrocyt nad 5,1 x /l, 10 % pak v rozmezí δ,51-5,00 x /l a pouze γ pacienti měli erytrocyt méně neţ 4,51 x /l (Tab. 14 a Tab. 15). Všichni vyšet ení měli rovněţ mikrocytózu (εcv < Ř0 fl) a hypochromii (MCH < 27 pg). Hodnota HbA 2 byla zvýšená u ř1 % pacient. V časopise Vnit ní léka ství v roce 1993 byla uve ejněna rozsáhlá studie československé kooperativní skupiny pod vedením Indráka (Indrák, 1řřγ), která shrnuje výsledky molekulárně-genetických vyšet ení u 135 osob z 5δ nep íbuzných rodin se suspektní talasemií z tehdejšího Československa. Vzorky pocházely z pracovišt z Prahy, Olomouce, Hradce Králové, Brna, Bratislavy, Banské Bystrice a Košic. εolekulárněgenetická vyšet ení byla provedena na pracovišti v USA v ůugustě (Medical College of Georgia) v laborato i prof. Huismana. Tato studie prokázala, ţe se na území bývalého 75
77 Československa vyskytuje poměrně široké spektrum talasemických alel bylo detekováno 1β r zných talasemických variant, z nichţ mutace IVS-I-1 (G-A), CD 39 (C- T), IVS-II-1 (G-A), IVS-I-110 (G-A) a IVS-II-745 (C-G) p edstavovaly 80 % všech talasemických mutací, které byly detekovány v té době v české a slovenské populaci, a k jejich rozší ení na našem území došlo z nějvětší pravděpodobnosti migrací obyvatelstva z mediteránních oblastí. Mutace IVS-I-1 (G-A) byla uváděná jako jedna z nejčastějších mediteránních mutací (Indrák, 1řřγ). Zbývající detekované mutace bylo moţné za adit mezi poměrně vzácné varianty a γ mutace nebyly d íve publikovány. εezi nově objevené mutace pat ily posunová mutace v CD 38/39 (-C), mutace v kodonu 121 (G-C) a mutace v kodonu 115 (GCC-GAC), která vedla k tvorbě vysoce nestabilního hemoglobinu - Hb Hradec Králové. V jedné rodině byla detekována rovněţ nová mutace v promotorové oblasti globinového genu (A-C), která byla p íčinou tzv. švýcarského typu nedelečního p etrvávání produkce fetálního hemoglobinu (HPFH). Tato forma HPFH bývá zp sobena mutacemi v promotorové oblasti jednoho nebo obou globinových gen, které vedou k zvýšené afinitě vazebných míst pro pozitivní transkripční faktory (Horváthová & Divoký, 2013). Po této rozsáhlé studii následovala další studie olomoucké skupiny uve ejněná v roce 2005 v časopise Vnit ní léka ství (Divoký et al., 2005). Z této studie vyplývá skutečnost, ţe v české a slovenské populaci jsou diagnostikování zejména heterozygoti pro + i 0 talasemii, tj. klinicky témě asymptomatičtí jedinci s mírně niţší hladinou Hb (hodnoty kolem 100 g/l), s mikrocytózou (εcv fl), hypochromázií a často i nápadnou erytrocytózu (aţ /l) odpovídající talasemii minor. Nejfrekventovanější mutací detekovanou v rámci souboru 183 pacient byla stejně jako v p edchozích studiích (Indrák, 1992, 1993 a 1994) mutace IVS-I-1 (G-A). Ve srovnání s touto poslední publikovanou analýzou talasemických alel v české a slovenské populaci se rozloţení četnosti jednotlivých mutantních alel v této prezentované práci mírně změnilo. Opět byly nejčastěji zastoupeny mutace mediteránního p vodu, a to zejména mutace IVS-I-110 (G-A) nově pak byly detekované mutace CD5β(-A) a CD41/42 (-TTCT) (Tab. 20). Obr. 21 srovnává výsledky β p edchozích studií (Indrák et al., 1993 Divoký et al., β005) a prezentované práce. Jednou z pravděpodobných p íčin 76
78 změny zastoupení jednotlivých talasemických alel je zvýšená migrace obyvatel, v menší mí e také zavedení sekvenačních analýz -globinového genu namísto p vodně pouţíváných restrikčních analýz, které umoţňovaly stanovit jen omezené mnoţství mutací vedoucí k vytvo ení/zrušení štěpného místa (p íkladem je nap. mutace IVSI-1(G-A)). talasemie major, která je charakteristická pro dvojité heterozygoty nebo homozygoty talasemické alely, se v České republice vyskytuje jen výjimečně. Tomu odpovídá i skutečnost, ţe všichni pacienti z vyšet ovaného souboru jsou heterozygotními nosiči mutace s klinickými projevy talasemie minor stejně tak jako v p edchozí studii. Obr. 21: Nejfrekventovanější zastoupení talasemických alel v české a slovenské populaci ze γ studií. Vzhledem ke zvyšující se migraci a s ní související zvyšující se incidenci talasemických alel u nás je t eba myslet na talasemii i v rámci prenatální diagnostiky; zvyšuje se totiţ pravděpodobnost výskytu talasemické alely u obou rodič s potencionální hrozbou pro výskyt dvojitého heterozygota resp. homozygota, který by byl postiţen -talasemií major. Beta talasemie lze odhalit nejen pomocí klasických invazivních metod prenatální diagnostiky; dnes je jiţ moţné vyšet ení fetální DNů z krve matky čímţ lze talasemii také rozpoznat. Bylo zjištěno, ţe v krvi matky je moţné zachytit volně 77
79 cirkulující DNů plodu (Lo et al., 1997). Tato metoda, známá od roku 1řř7, je však zatím poměrně finančně náročná pro rutinní diagnostiku. I kdyţ primárně byla tato práce zamě ená na charakterizaci výskytu talasemické alely v české populaci, v menší mí e se zabývá rovněţ identifikací p íčiny methemoglobinémie u γ pacient a potvrzením/vyvrácením koincidence primární hemochromatózy u pacient s deficitem pyruvátkinázy. Vrozená methemoglobinémie typu I a II je autosomálně recesivní onemocnění, které je spojováno s deficitem enzymu cytochrom-b5-reduktázy podílejícím se na zpětně redukci methemoglobinu (kap. 4.1). Oba typy je nutné odlišit zejména od další vrozené methemoglobinémie, která je autosomálně dominantního charakteru a je výsledkem výskytu hemoglobinu ε (kap. 4.2). U 2 pacient byla detekována sníţená aktivita cytochrom-b5-reduktázy (β5-γ0 % kontrolních aktivit). Na molekulární úrovni bylo prokázáno, ţe všichni pacienti jsou heterozygoti pro mutaci v genu CYB5R3 kódující tento enzym a to mutaci c.400c G, c.890g A a c.316g ů. Všechny mutace se nacházejí ve FAD-vazebné doméně. Mutace c.400g C a c.316g ů se nacházejí v oblasti, která interaguje s NAD(P)H vazebnou doménou enzymu. P estoţe se ve všech p ípadech jedná o jednoduché heterozygoty s mírnou resp. asymptomatickou methemoglobinémií (εethb 11 %), není moţné kauzalitu v recesivní konfiguraci jednoznačně hodnotit. V české populaci zatím nebyly popsány p ípady methemoglobinémie typu I ani II a to u dvojitých heterozygot nebo homozygot pro mutaci v genu CYB5R3, ani p ípady methemoglobinémie související s deficitem enzymu cytochrom-b5-reduktázy. Výskyt methemoglobinémie enzymového typu je většinou endemického charakteru vysoká frekvence jejího výskytu je mezi p vodními obyvateli ůljašky a rovněţ mezi indiány kmene Navajo (Scott, 1960 Balsamo et al., 1964). U pacient s deficitem pyruvátkinázy, stejně tak jako u pacient s talasemií, často dochází k sekundárnímu p etíţení organizmu ţelelezem (kap. 5). P íčinou m ţe být celá ada faktor chronická hemolýza, inefektivní erytropoéza, splenektomie, transfuzní terapie pop. koincidence s primární hemochromatózou (Zanella et al., 1993). V rámci studovaného souboru, devět z jedenácti pacient mělo zvýšené hodnoty feritinu a to včetně 78
80 pacienta, který nikdy nepodstoupil transfuzní terapii. Pět z nich mělo rovněţ zvýšenou saturaci transferinu. Efekt splenektomie nebylo moţné jednoznačně hodnotit. Splenektomování byli pouze β pacienti, v jednom p ípadě došlo k zvýšení hodnot feritinu, v druhém naopak k sníţení. Vzhledem k tomu, ţe druhý pacient je dítě i tak s poměrně vysokými hodnotami feritinu (cca β000 µg/l) (Tab. 22), je moţné, ţe se efekt splenektomie projeví teprve s časem. Sekvenační analýza genu HFE u těchto pacient potvrdila abnormální genotyp určující predispozici k p etíţení ţelezem (Zanella et al., 2001) u 4 z nich (Tab. 23). Jediný (dětský) pacient, který je dvojitým heterozygotem pro mutaci p.cysβřβtyr a p.his6γůsp, však nevykazoval zvýšené hodnoty feritinu. Koincidenci s primární hemochromatózou stejně tak jako v p ípadě vlivu splenektomie, nebylo moţné jednoznačně hodnotit. Protoţe se primární hemochromatóza typu 1 projevuje zejména aţ po γ. dekádě ţivota (Pietntragello, 2010), je moţné, ţe se její vliv projeví u tohoto pacienta aţ v jeho dospělosti. Ani u pacienta s hyperferitinémií nebyla potvrzena kauzální mutace pro hemochromatózu typu I, II, III ani IV. Bylo u něj detekováno 5 SNP variant v genu TFR2, SLC40A1 a FTL, z nichţ c.δδ-24g C je sice v literatu e spojována se zhoršením projev hemochromatózy typu I (ůltès et al., β00ř), nicméně tento SNP byl detekován rovněţ u všech ostatních pacient s deficitem pyruvátkinázy, aniţ by se projevil jeho efekt. Ukázalo se však, ţe chelatační terapií s prodluţováním interval mezi jednotlivými transfuzemi, které jsou podle všeho jednou z hlavních p íčin p etíţení ţelezem u pacient s deficitem pyruvátkinázy, došlo u tohoto pacienta v jeho cca 2,5 letech k výraznému poklesu feritinu z cca δ000 na 1δ5 µg/l (referenční rozmezí pro dětské pacienty je 70 µg/l (Brugnara, 2009)). Z tohoto lze usuzovat, ţe jeho těţký novorozenecký fenotyp je výsledkem deficitu pyruvátkinázy homozygotní mutace p.ůrg51řfs v genu PKLR zp sobuje posun čtecího rámce a tvorbu proteinu, který má o δ6 aminokyselin kratší etězec enzymu chybí část aktivační domény včetně aktivačního místo ůrg5γβ umoţňjící vazbu p irozeného aktivátoru fruktóza-1,6-bisfosfátu. Jedná se tak o první p ípad popsané neonatální hyperferitinémie asociované s deficitem pyruvátkinázy. 79
81 11. Závěr Tato rigorózní práce shrnuje výskyt talasemických mutací HBB genu v české i slovenské populaci u 1Řγ pacient ze 10δ nep íbuzných rodin, kte í byli do studie zahrnuti v letech Navazuje tak jiţ na d íve publikované studie. Stejně jako v p edchozích p ípadech potvrzuje výskyt všech talasemických mutací v heterozygotní formě s p evaţujícím fenotypovým projevem talasemie minor vyjma mutace CD121(G- T) tj. c.364g>t vedoucí k p edčasné terminaci translace projevující se st edně těţkou aţ těţkou anémií s obrazem talasemie intermedia. Naše výsledky se však liší frekvencí výskytu jednotlivých mutací, coţ je s největší pravděpodobností dáno zvýšenou migrací obyvatelstva. Celkem bylo identifikováno 15 r zných exonových nebo intronových mutací globinového genu, jedna krátká delece δδ nukleotid a t i u nás méně časté polymorfismy RS713040, RS a RSγδ0βřγř0, u kterých však souvislost s talasemickým fenotypem zatím nebyla jednoznačně potvrzena. Nejčastěji se vyskytovaly tzv. sest ihové mutace mediteránního p vodu IVS-I-110(G-A), CD39(C-T), IVS-I-1(G- A) a IVS-I-6(T-C) resp. c-93-21g A, c.118 T, c.92+1g A a c.92+6t C (s frekvencí výskytu v souboru 10 %). V rámci souboru byly detekovány i mutace, které nebyly zatím na území bývalého Československa prokázány, jednalo se o mutaci CD52(-A) a CD41/42(- TTCT) tj. c.52dela a c.126_129delcttt. Druhá z těchto mutací je asijského p vodu a byla nalezena u imigranta z Vietnamu. Dále bylo v rámci této práce na souboru γ pacient s mírnou resp. asymptomatickou methemeoglobinémií potvrzeno, ţe všichni pacienti mají sníţenou aktivitu enzymu cytochrom-b5-reduktázy (cca β5 % kontrolních hodnot) a jsou heterozygoti pro mutaci v genu CYB5R3 (c.316g A, c.400c G a c.890g ů). Nicméně vlivem recesivní konfigurace nelze methemoglobinémií blíţe u těchto pacient klasifikovat. Na souboru pacient s deficitem pyruvátkinázy bylo prokázáno, ţe feritinémie u těchto pacient m ţe vzniknout jak u chronicky transfundovaných pacient, tak i nezávisle na transfuzich. Vliv splenektomie stejně jako koincidence s vrozenou hemochromatózou nebylo naopak u těchto pacient moţné jednoznačně hodnotit a to zejména vlivem jejich nízkého věku. Rovněţ u pacienta s hyperferitinémií nebyla prokázána koincidence s primární hemochromatózou vyloučením kauzální mutace v genech pro hemochromatózu typu I aţ IV. Úpravou feritinémie chelatační terapií bylo naopak potvrzeno, ţe jeho těţký 80
82 novorozenecký fenotypový projev je výsledkem deficitu pyruvátkinázy. Jedná se tak o v bec první dokumentovaný p ípad hyperferitinémie asociované s deficitem pyruvátkinázy. Tato práce p ispěla k identifikaci nových talasemických mutací na našem území a k získání nových informací o rozší ení talasemických alel zejména s p ihlédnutím k nár stu migrace obyvatelstva, ke zlepšení diagnostiky methemoglobinémie enzymového charakteru a rovněţ k bliţšímu pochopení souvislosti mezi p etíţením ţelezem a hemolytickou anémií v d sledku deficitu enzymu pyruvátkinázy. 81
83 SEZNAM OBRÁZKŮ Obr. 1: Zjednodušené schéma hematopoézy Obr. 2: Struktura hemoglobin a hemu Obr. γ: Schematické znázornění α globinového lokusu na chromozomu 16 a globinového genu na chromozomu 11 (ů). Exprese globinového genu (B) Obr. δ: Syntéza globinových etězc u lidského plodu a kojence Obr. 5: P íklady bodových mutací globinového genu zp sobujících talasemii Obr. 6: Redukce methemoglobinu na hemoglobin prost ednictvím cytochrom-b5- reduktázou Cb5R NADH Obr. 7: Krystalová struktura proteinu Cb5R s FAD a NAD(P)H-vazebnou doménou Obr. 8: Mutace IVS-I-110 (G-A) Obr. 9: Mutace CD39 (C-T) Obr. 10: Mutace IVS-I-1 (G-A) Obr. 11: Mutace IVS-I-6 (T-C) Obr. 12: Mutace IVS-II-1 (G-A) Obr. 1γ: Posunová mutace v CD Ř (-AA) Obr. 1δ: Posunová mutace CD γř/γř (-C) Obr. 15: Mutace CD 52 (-A) Obr. 16: Mutace CD 41/42 (-TTCT) Obr. 17: 3 polymorfismy u jednoho pacienta Obr. 18: Mutace v genu CYB5R Obr. 19: ůminokyselinová sekvence lidské erytrocytární NůDH-Cb5R Obr. 20: Pacient s p etrávající hyperferitinémií Obr. 21: Zastoupení nejfrekventovanějších talasemických alel
84 SEZNAM TABULEK Tab. 1: Rozdělení vrozených hemochromatóz Tab. β: Sloţení PCR reakční směsi Tab. γ: Sekvence pouţitých primer pro amplifikaci exon genu HBB Tab. δ: Sekvence pouţitých primer pro amplifikaci exon genu CYB5R Tab. 5: Sekvence pouţitých primer pro amplifikaci exon genu HFE Tab. 6: Sekvence pouţitých primer pro amplifikaci exon genu HFE2 a HAMP Tab. 7: Sekvence pouţitých primer pro amplifikaci exon genu TFR Tab. Ř: Sekvence pouţitých primer pro amplifikaci exon gen SLC40A1 a FTL Tab. ř: Časový a teplotní profil PCR reakce - amplifikace úsek genu HBB Tab. 10: Časový a teplotní profil PCR reakce - amplifikace úsek genu CYB5R3 a gen pro HH Tab. 11: Sloţení reakční směsi pro cyklické sekvenování Tab. 1β: Koncentrace produktu pro sekvenační reakci dle velikosti PCR produktu Tab. 1γ: Časový a teplotní profil sekvenační reakce Tab. 14: Počet erytrocyt u muţ Tab. 15: Počet erytrocyt u ţen Tab. 16: Hodnoty hemoglobinu u muţ Tab. 17: Hodnoty hemoglobinu u ţen Tab. 18: Hodnoty HbA 2 u muţ i ţen Tab. 19: Hodnoty hemoglobinu F v souboru pacient Tab. β0: talasemické mutace detekované v české a slovenské populaci u 1Řγ pacient ze 105 rodin Tab. 21: Pacienti se sníţenou aktivitou Cb5R biochemická a molekulárně-genetická charakterizace Tab. ββ: Klinická data pacient s deficitem pyruvátkinázy Tab. 23: Mutace v genu PKLR a HFE u pacient s deficitem pyruvátkinázy
85 SEZNAM POUŽITÝCH ZKRATEK 2,3-BPG 2,3-bifosfoglycerát A adenin ATP adenosine triphosphate, adenosintrifosfát BFU-E Burst Forming Unit-Erythroid, prekurzor erytrocyt Bp base pair, počet nukleotid C cytosin CD36 Cluster of differentiation, diferencuiační antigen CFU Colony Forming Unit, buňka tvo ící kolonie, progenitorová kmenová buňka CFU-E Colony forming Unit-Erythroid, progenitorová kmenová buňka erytrocyt CFU-Eos Colony Forming Unit Eosinophil, pro buňky eosinofi CFU-G Colony Forming Unit Granulocyte, pro buňky granulocytární ady CFU-GEMM Colony Forming Unit Granulocyte Erytrocyte Macrophage Megakaryocyte, myeloidní kmenová buňka CFU-GM Colony Forming Unit Granulocyte Macrophage, bipotentní kmenová buňka pro makrofágovou a granulocytární adu CFU-M Colony Forming Unit Monocyte, pro buňky monocytární ady CFU-Meg Colony Forming Unit Megakaryocyte, pro buňky megakaryocytové ady Da Dalton, jednotka relativní molekulové hmotnosti DNA Deoxyribonucleic acid, deoxyribonukleová kyselina DTT dithiothreiotol EB Elution buffer, eluční pufr EDTA Ethylendiamintetraoctová kyselina EKLF Erythroid Kruppel-Like Factor, erytroidní transkripční faktor EPO Erythropoetin, erytropoetin FAD flavinadenindinukleotid G guanin G6PD Glucose-6-phosphate dehydrogenase, glukóza-6-fosfátdehydrogenáza Hb hemoglobin HbA hemoglobin dospělého typu HbF fetální hemoglobin 84
86 Hct hematokrit HPFH Hereditary persistence of fetal hemoglobin, dědičné p etrvávání fetálního hemoglobinu IVS intron Kb kilobáze LB Loading buffer, pufr pro nanášení vzork LCR Locus Control Region MCV Mean Corpuscular Volume, st ední objem erytrocytu MCH Mean Corpuscular Hemoglobin, pr měrné mnoţství hemoglobinu v erytrocytu MCHC Mean Corpuscular Hemoglobin Concentration, st ední koncentrace hemoglobinu v erytrocytu mrna Messenger RNA, mediátorová ribonukleová kyselina NADH redukovaná forma nikotinamidadenindifosfonukleotidu NaOH hydroxid sodný (NH 4 ) 2 SO 4 síran amonný PBS Phosphate buffer saline, fosfátový pufr PCR Polymerase Chain Reaction, polymerázová etězová reakce PE Wash buffer, promývací pufr QG Solubilization buffer, vyrovnávací pufr RBC Red blood cells, erytrocyty RDW Red blood cell distribution width, distribuční ší e erytrocyt RNA Ribonucleic acid, ribonukleová kyselina RT Room temperature, pokojová teplota SDS Sodium dodecyl sulfate, dodecylsíran sodný STOP terminační kodón TBE Tris/Borát/EDTů pufr Tris Tris(hydroxymethyl)aminomethan 85
87 SEZNAM LITERATURY Allen S. J., O Donnell A., Alexander N. D. E., Alperts M. P., Peto T. E. A., Clegg J. B., Weatherall D. J. (1997): α -thalassemia protects childrenagainst desease caused by other infections as well as malaria. Proceedings of the National Academy of Science USA 94: Altès A., Bach V., Ruiz A., Esteve A., Remacha A. F., Sarda M. P., Felez J., Baiget M. (2009): Does the SLC40A1 gene modify HFE-related haemochromatosis phenotypes? Annals of Hematology, 88: Badens C., εattei ε. G., Imbert ů. ε., δapouméroulie C., εartini N., εichel G., δena- Russo D. (2002): A novel mechanism for thalassaemia intermedia. Lancet 359(9301): Balsamo, P., Hardy, W. R., Scott, E. M. (1964): Hereditary methemoglobinemia due to diaphorase deficiency in Navajo Indians. Journal of Pediatrics 65: Barbour V. M., Tufarelli C., Sharpe J. A., Smith Z. E., Ayyub H., Heinlein C. A., Sloane- Stanley j., Indrák K., Wood W. G., Higgs D. R. (β000): Alpha-thalassemia resulting from a negative chromosomal position effect. Blood 96: Baysal E., Indrak K., Bozkurt G., Berkalp A., Aritkan E., Old J. M., Ioannou P., ůngastiniotis ε., Droushiotou ů., Yuregir G. T., Kilinç Y., Huisman T. H. J. (1řřβ): The beta-thalassaemia mutations in the population of Cyprus: British Journal of Haematology 81: Beard J. L. (2001): Iron biology in immune function, muscle metabolism and neuronal functioning. The Journal of nutrition 131(2S-2): 568S-579S; discussion 580S Beard, J., Han O. (2009): Systemic iron status. Biochemica et Biophysica Acta 1790(7): Bewley M. C., Marohnic C. C., Barber M. J. (2001): The structure and biochemistry of NADH-dependent cytochrome b5 reductase are now consistent. Biochemistry. 40 (45): Bilgen T., Canatan D., Ankan. Y., Yesilipek A., Keser I. (2011): The effect of HBB:c.*+96T > C (3 ' UTR+1570 T > C) on the mild beta-thalassemia intermedia phenotype. Turkish Journal of Hematology. 28(3): Boyer S. H., Bishop T. R., Rogers O. C., Noyes A. N., Frelin L. P., Hobbs S. (1992): Roles of erythropoietin, insulin-like growth factor 1, and unidentified serum factors in promoting maturation of purified murine erythroid colony-forming units. Blood 80: Brabec V. (1988): Beta-talasemie v českých rodinách. Vnit ní léka ství γδ: Brugnara C. (2009): Reference values in infancy and childhood. In: Nathan and Oskiʼs hematology of infancy and childhood (7th ed.). Saunders elsevier. Philadelphia
88 Cai S. P., Eng B., Francombe W. H., Olivieri N. F., Kendall A. G., Waye J. S., Chui D. H. (1992): Two novel beta-thalassemia mutations in the 5' and 3' noncoding regions of the beta-globin gene. Blood 79(5): Camaschella C., Roetto A., Calì ů., De Gobbi M., Garozzo G., Carella M., Majorano N., Totaro A., Gasparini P. (2000): The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nature Genetics, 25(1): Cao A., Rosatelli M. C., Eckman J. R. (2001): Prenatal diagnosis and screening for thalassemia and sickle cell disease. In: Disorders of haemoglobin: Genetics, pathophysiology and clinical management. Ed.: Steinberg M. H., Forget B. G., Higgs D. R., Nagel R. L. Cambridge University Press. Cambridge Catlin A. J. (2003): Thalassemia: the facts and the controversies. Pediatric Nursing 29(6): Češka R. (β010): Interna. Praha: Triton, 1. vydání, Ř55 s. ISBN: Chrobák δ. (β001): Mikrocytární a hypochromní anémie. Vnit ní léka ství δ7(γ): Čimburová ε., P tová.i, Provazníková H., Pintérová D., Horák J. (β005): S65C and other mutations in the haemochromatosis gene in the czech population. Folia Biologica 51(6): Clark B. E., Thein S. L. (2004): Molecular diagnosis of haemoglobin disorders. Clinical and Laboratory Haematology 26(3): Clegg J. B., Metaxatou-Mavromati A., Kattamis C., Sofroniadou K., Wood W. G., Weatherall D. J. (1979): Occurrence of G gamma Hb F in Greek HPFH: analysis of heterozygotes and compound heterozygotes with beta thalassaemia. British Journal of Haematology 43(4): Cole S. K., Billewicz W. Z., Thomson A. M. (1971): Sources of variation in menstrual blood loss. Journal of Obstetrics and Gynaecology of the British Commonwealth 78 (10): Cunningham, M. J., Macklin E. A., Neufeld E. J., Cohen A. R. (2004): Complications of beta-thalasemia major in North America. Blood 104(1): Deisseroth A., Nienhuis A., Lawrence J., Giles R., Turner P., Ruddle F. H. (1978): Chromosomal localization of human β globin gene on human chromosome 11 in somatic cell hybrids. Proc Natl Acad Sci U S A. 75(3): Dickerson R. E., Geis I. (1983): Hemoglobin: structure, function, evolution, and patology. Ed.: C. A. Benjamin. Cummings Publishing Co., Menlo Park, 176 p. ISBN Divoký V., Baysal E., Öner R., Çürük M. A., Walker E. δ. D., Indrák K., Huisman T. H. J. (1994): The T-C mutationat position +96 of the untranslated region 3 to the terminating codon of the β-globin gene is a rare polymorfism that does not cause a β-thalassemia as previously ascribed. Human Genetics 93(1): Divoký V., Walczyskova S., Pospíšilová D., Priwitzerová ε., Takáčová S., Kostelecká I., Divoká ε., Roţmanová Š., Jarošová ε., Čermák J., Indrák K. a Česko-slovenská kooperativní skupina pro diagnostiku talasemií (β005): Některé vzácnější formy hereditárních anémií vyskytující se v dospělé populaci v ČR a SR β talasemie a nestabilní hemoglobinové varianty. Vnit ní léka ství 51: ŘŘ
89 Divoký V., Indrák K., εojzíková R. (β01γ): Hemoglobinopatie:talasémie a strukturální Hb varianty. In: εolekulární hematologie (1. vydání). Pospíšilová Š. et al. (Eds.). Galén. Praha. β Eng B., Waye J. S., Chui D. H. K. (1992): The T-C substitution at nucleotide of the beta-globin gene is a polymorphism. Blood 80(5): Fábryová V., Drakulová ε., Sakalová ů. (2004): Výskyt beta-talasémií na Slovensku. δekársky Obzor 53(7-8): Fábryová V., Sakalová ů. (β006): Novosti v diagnostike, liečbe a prevencii beta-talasémií. δekársky Obzor 55(10): Fábryová V. (β007): Beta-talasémie. Bratislava: Herba, 1. vydání, řβ s. ISBN Feder J.N.,Gnirke A, Thomas W., Tsuchihashi Z., Ruddy D. A., Basava A., Dormishian F., Domingo R. Jr, Ellis M. C., Fullan A., Hinton L. M., Jones N.L.,Kimmel B.E.,Kronmal G.S., Lauer P., Lee V.K., Loeb D. B., Mapa F. A., McClelland E., Meyer N. C., Mintier G. A., Moeller N., Moore T., Morikang E., Prass C. E., Quintana L., Starnes S. M.,Schatzman R. C., Brunke K. J., Drayna D. T., Risch N. J., Bacon B. R., Wolff R. K. (1996): A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nature Genetics, 13(4): Forget B. G. (1998): Molecular basis of hereditary persistence of fetal hemoglobin. Annals of the New York Academy of Sciences 850: Frassinelli-Gunderson E. P., Margen S., Brown J. R. (1985): Iron stores in users of oral contraceptive agents. The American Journal of Clinical Nutrition 41(4): Galanello R., Origa R. (2010): Beta-thalassemia. Orphanet Journal of Rare Diseases, 5: 11. Ganz T. (2004): Hepcidin in iron metabolism. Current opinion in hematology 11(4): Harteveld C. L., Osborne C. S., Peters M., van der Werf S., Plug R., Fraser P., Giordano P. C. (2003): Novel 112 kb (epsilonggammaagamma) deltabeta-thalassaemia deletion in a Dutch family. British journal of haematology 122(5): Higgs D. R., Engel J. D., Stamatoyannopoulos G. (2012): Thalassaemia. The Lancet 379(9813): Hlobilová ε. (β011): εolekulárně genetická analýza vybraných hematologických chorob. Diplomová práce. Olomouc: Univerzita Palackého. P írodověděcká fakulta. 106s. Vedoucí diplomové práce: RNDr. Martina Divoká Hoffbrand A. V., Pettit J. E. (1988): Clinical Hematology. Sandoz Atlas. (1st edition). Gower Medical Publishing. London Horváthová ε., Pospíšilová D. (β010): Nové poznatky o homeostaze železa a jejich důsledky pro klinickou praxi. Postgraduální medicína 1β: Horvathova M., Kapralova K., Zidova Z., Dolezal D., Pospisilova D., Divoky V. (2012): Erythropoietin-driven signaling ameliorates the survival defect of DMT1-mutant erythroid progenitors and erythroblasts. Haematologica. 97(10):
90 Horvathová ε., Divoký, V. (β01γ): Metabolismus železa a jeho poruchy. In: εolekulární hematologie (1. vydání). Pospíšilová Š. et al. (Eds.). Galén. Praha. β Hunt, J. R. Iron, USDA-ARS Grand Fork Human nutrition research center, 2005, Indrák K., Indráková J., Kultar F., Pospíšilová D., I. Sulovská E., Baysal E., Huisman T. H. (1991): Compound heterozygosity for a beta zero-thalassemia (frameshift codons 38/39; -C) and a nondeletional Swiss type of HPFH (A----C at NT -110, G gamma) in a Czechoslovakian family. Annals of Hematology 63(2): Indrák K., Brabec V., Indráková J., Chrobák δ., Sakalová ů., Jarošová ε., Čermák J., Fei Y. J., Kutlar F., Gu Y. Ch., Baysal E., Huisman T. H. J. (1992): Molekular characterization of β-thalassemia in czechoslovakia. Human Genetics 88(4): Indrák K. (1řřγ): Hemoglobinopatie v České a Slovenské populaci se zaměřením na problematiku β-talasemií a jejich molekulárně genetickou identifikaci. Dizertační práce. Univerzita Karlova v Praze. 168 s. Indrák K., Divoký V., Brabec V., Indráková J., Svobodová ε., Huisman T. H. (1řřγb): Molekulárně genetická charakteristika alfa, beta a deltabeta thalassémií u 139 heterozygotů z 56 nepříbuzných rodin českého a slovenského původu. Vnit ní léka ství γř(10): ř6ř-978. Indrák K., Divoký V., Brabec V., Chrobák δ., εociková K., Sakalová ů., Svobodová ε., Indráková J., Hammerová T., Zarnovicanová ε. (1řřδ): Dominantní β-talasemické alely v českéa slovenské populaci [β-talasemické mutace v 112 (T-A) a 121 (G-T) a nestabilní hemoglobinová varianta Hradec Králové nebo α2β2115 (Gl7) Ala-Asp]. Vnit ní léka ství δ0(δ): ββγ 230. Indrák K., Divoká ε., εelichárková R., δuhový ε., Divoký V. (1řřŘ): Hemoglobin Haná nebo alpha 2 beta 2 63 (E7) His-Asn: nová nestabilní varianta hemoglobinu s paradoxnĕ rozdílnou klinickou manifestací u kuráků a nekuráků téze rodiny. Vnit ní léka ství 43(5): Jaffe E. R. (1986): Enzymopenic hereditary methemoglobinemia: a clinical/biochemical classification. Blood Cells 12: Kaushansky K. (2009): Determinants of platelet number and regulation of thrombopoiesis. Hematology. American Society of hematology. Education Program Kazazian H. H. Jr., Dowling C. E., Hurwitz R. L., Coleman M., Stopech A., Adams J. G. 3rd. (1992): Dominant thalassemia-like phenotypes associated with mutations in exon 3 of the beta-globin gene. Blood 79(11): Ketley N. J., Newland A., C. (1997): Haemopoietic growth factors. Postgraduate Medical Journal 73(858): Klener P., Cieslar P., Friedmann B., Šálková J., Trněný ε. (β00γ): Hematologie. 1. vydání. Praha: Galén, 115 s. ISBN Koury M. J., Bondurant M. C. (1990): Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science 248(4953):
91 Krahulcová E., εatýšková ε., Penka ε. (1řř6): Hematologie: pro zdravotní sestry na transfúzních odděleních. První. Brno: Institut pro další vzdělávání pracovník ve zdravotnictví, 1γ1 s. ISBN Kynclová E., Kovaríková δ., Fajkošová P., εelichárková R., Indráková J., Brabec V., Čermák J., Indrák K. (1řřŘ): Haplotypy beta-globinového lokusu Čechů a slováků s ebta-talasemiemi a strukturními variantami hemoglobinu. Vnit ní léka ství δδ(ř): Kynclová E., Divoký V., Kovaríková L., εelichárková R., Indráková J., Divoká M., Hammerová T., Sakalová A., Hudecek J., Indrák K. (1999): New beta0- thalassaemic insertion mutation (CD 7/8, +G) in a Slovak family, associated with the Mediterranean haplotype IX. Vnit ní léka ství 45(3): Lichtman M. A., Williams W. J. (2006): Williams hematology. (7th edition). McGraw- Hill, Medical Pub. Division. New York Lo Y. M., Chan K. C., Sun H., Chez E. Z., Jiang P., Lun F. M., Zheng Y. W., Leung T. Y., Lau T. K., Cantor C. R., Chiu R. W. (2010): Maternal Plasma DNA Sequencing Reveals the Genome-Wide Genetic and Mutational Profile of the Fetus. Science translational medicine 2(61): Lukens J. N. (1999): The thalassemia and related disorders, quantitative disorders of hemoglobin synthesis In: Wintrobe s clinical hematology. (10th edition). Lea + Fiebeger. London Mansouri A., Lurie A. A. (1993): Concise review: methemoglobinemia. American Journal of Hematology. 42(1): Melchers F., Rolink A. (2001): Hematopoietic Stem Cells: Lymphopoiesis and the Problem of Commitment Versus Plasticity. In: Marshak D. R., Gardner R. L., Gottlieb D. I. (ed.): Stem cell biology. Cold Spring Harbor, Cold Spring Harbor Laboratory Press. New York Montosi G., Donovan A., Totaro A., Garuti C., Pignatti E., Cassanelli S., Trenor C. C., Gasparini P., Andrews N. C., Pietrangelo A. (2001): Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. The Journal of Clinical Investigation. 108(4): Muirhead N., Bargman J., Buregess E., Jindal K. K., Levin A., Nolin L., Parfrey P. (1995): Evidence-Based recommendations for the clinical use of recombinant human erythropoietin. American Journal of Kidney Diseases 26(2 Suppl 1): Neishabury M., Azarkeivan A., Oberkanins C., Esteghamat F., Amirizadeh N., Najmabadi H. (2008): Molecular mechanisms underlying thalassemia intermedia in Iran. Genetic Testing 12(4): Nienhuis A. W., Stamatoyannopoulos G. (1978): Hemoglobin switching. Cell: Nienhuis A. W., Anagnou N. P., Ley T. J. (1984): Advances in thalassemia research. Blood 63: Nuntakarn L., Fucharoen S., Fucharoen G., Sanchaisuriya K., Jetsrisuparb A., Wiangnon S. (2009): Molecular, hematological and clinical aspects of thalassemia major and 90
92 thalassemia intermedia associated with Hb E-beta-thalassemia in Northeast Thailand. Blood Cells, Molecules & Diseases 42(1): Nussbaum R. L. McInnes R. R., Huntington F. W. (2004): Klinická genetika: Thompson & Thompson. Šesté. Praha: Triton, δβ6 s. ISBN Orkin S. H., Zon L. I. (2002): Hematopoiesis and stem cells: plasticity versus developmental heterogenity. Nature Imunology 3(4): Palstra R. J., de Laat W., Grosveld F. (2008): Beta-globin regulation and long-range interactions. Advances in Genetics 61: Papanikolaou G., Samuels M. E., Ludwig E. H., MacDonald M. L., Franchini P. L., Dubé M. P., Andres L., MacFarlane J., Sakellaropoulos N., Politou M., Nemeth E., Thompson J., Risler J. K., Zaborowska C., Babakaiff R., Radomski C. C., Pape T. D., Davidas O., Christakis J., Brissot P., Lockitch G., Ganz T., Hayden M. R., Goldberg Y. P. (2004): Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nature Genetics. 36(1): Papanikolaou G., Tzilianos M., Christakis J. I., Bogdanos D., Tsimirika K., MacFarlane J., Goldberg Y. P., Sakellaropoulos N., Ganz T., Nemeth E. (2005): Hepcidin in iron overload disorders. Blood 105(10): Pecka M. (2002): Laboratorní hematologie v přehledu I. Buňka a krvetvorba. Český Těšín: FINIDR, 1. vydání, 160 s. ISBN Penka M., Buliková ů., εatýšková ε. (2001): Hematologie I: Neonkologická hematologie. 1. vydání. Praha: GRůDů Publishing β1δ s. ISBN Penka ε., Buliková ů., εatýšková ε., Novotný J., Ko ístek Z., Bourková δ., Zav elová J., Čech Z., Snopková S. (β00ř): Neonkologická hematologie: 2., doplněné a zcela přepracované vydání. β. vydání. Praha: Grada Publishing, βδř s. Percy M. J., Gillespie M. J. S., Savage G., Hughes A. E., McMullin M. F., Lappin T. R. J. (2002): Familial idiopathic methemoglobinemia revisited: original cases reveal 2 novel mutations in NADH-cytochrome b5 reductase. Blood 100: Percy M. J., Lappin T. R. (2008): Recessive congenital methaemoglobinaemia: cytochrome-b5-reductase deficiency. British Journal of Haematology. 141: Pietntragello A. (2010): Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterlogy, 139: , 408.e1-2. Ponka P. (1997): Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood 89(1): Popovich B. W., Rosenblatt D. S., Kendall A. G., Nishioka Y. (1986): Molecular characterization of an atypical beta-thalassemia caused by a large deletion in the 5' beta-globin gene region. American Journal Of Human Genetics. 39(6): Pospíšilová P. (β01β): Biochemická charakterizace enzymopatií způsobujících vrozené hemolytické anémie. Diplomová práce. Olomouc: Univerzita Palackého. P írodověděcká fakulta. 106s. Vedoucí diplomové práce: εgr. εojzíková Renáta, Ph.D. Prchal J. T., Gregg X. T. (2005): Red cell enzymes. American Society of Hematology Education Program 2005:
93 Prchal J. T., Gregg X. T (2000): Red cell enzymopathies. In: Hoffman R., Benz E. J. Jr, Shattil S. J., et al, eds. Hematology: basic principles and practice. (3rd ed). NY: Churchill Livingstone. New York Pr ša R. (1997): Základy analytických metod v klinické molekulární biologii. 1. vydání. Praha: δéka ská fakulta Univerzity Karlovy a δůεbdů BIO εed spol. s. r. o., δ5 s. ISBN Ringelhann B., Szelenyi J. G., Horanyi M., Svobodová M., Divoký V., Indrák K., Hollân S., Marosi A., Laub M., Huisman T. H. (1993): Molecular characterization of betathalassemia in Hungary. Human Genetics 92(4): Roetto A., Papanikolaou G., Politou M., Alberti F., Girelli D., Christakis J., Loukopoulos D., Camaschella C. (2003): Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nature Genetics 33(1): Sakalová ů., Hammerová t., Divoká ε., Indráková J., Kovaríková δ., εelichárková R., Divoký V., Kynclová E., Hudecek J., Indrák K. (1řřř): Nová beta0-talasemická posunová mutace (CD 7/8, +G) ve slovenské rodinĕ asociovaná s mediteránním haplotypem IX. [[New beta0-thalassaemic insertion mutation (CD 7/8, +G) in a Slovak family, associated with the Mediterranean haplotype IX]]. Vnit ní δéka ství 45(3): Scott, E.M. (1960): The relationship of diaphorase of human erythrocytes to inheritance of methemoglboinemia. J. Clin. Invest. 39: Sharpless N. E., DePinho R. A. (2007): How stem cells age and why this makes us grow old. Nature Reviews Molecular Cell Biology 8(9): Shear H. L, Grinberg L., Gilman J., Fabry M. E., Stamatoyannopoulos G., Goldberg D. E., Nagel R. L. (1998): Transgenetic mice expressinghuman fetal globin are protected from malaria by a novel mechanism. Blood 92(7): Stamatoyannopoulos G., Majerus P. W., Perlmutter R. M., Varmus H. (2001): The molecular basis of blood diseases (3rd ed.), WB Saunders Company, Philadelphia Štaud F. (2007): Molekulární farmakologie látek stimulujících erytropoézu. Remedia 17: Steinberg M. H., Forget B. G., Higgs D., Nagel R. L., Weatherall D. J. (2009): Disorders of hemoglobin: Genetics, Pathophysiology and Clinical management. (2nd) Cambridge University Press. Cambridge Thein S. L. (2004): Genetic insights into the clinical diversity of beta thalassaemia. British Journal of Haematology 124(3): Weatherall D. J. (2001): Phenotype-genotype relationships in monogenetic disease: lessons from the thalasseamias. Nature reviews. Genetics 2(4): Weatherall D. J. (2006): Disorders of globin sythesis: The thalassemias. McGraw-Hill Medical. USA Weatherall D. J., Lichtman M. A., Beutler E., Kipps T. J., Selingsohn U., Kaushansky K., Prchal J. T. (2006): Williams HEMATOLOGY. (7th edition). McGraw-Hill Medical. USA
94 Weatherall D. J. (2010): The inherited diseases of hemoglobin are an emerging global health burden. Blood 115(22): Weissman I. L., Anderson D. J., Gage F. (2001): Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annual review of cell and developmental biology 17: Yaish H. M., (2013): Pediatric thalassemia. Published online: Zanella A., Berzuini A., Colombo M. B., Guffanti A., Leccgu L., Poli F., Cappellini M. D, Barosi G. (1993): Iron status in red cell pyruvate kinase deficiency: Study of Italian cases. British Journal of Haematology, 83: Zanella A., Bianachi P., Iurio A., Boschetti C., Taioli E., Verllati C., Zappa M., Fermo E., Tavazzi D., Sampietro M. (2001): Iron status and HFE genotype in erythrocyte pyruvate kinase deficiency: study of Italian cases. Blood Cells Molecules and Diseases, 27:
95 SEZNAM PŘδLOH Příloha 1: Hemochromatóza (zahraniční publikace s IF). Mojzikova, R., Koralkova, P., Holub, D., Zidova, Z., Pospisilova, D., Cermak, J., Striezencova Laluhova, Z., Indrak, K., Sukova, M., Partschova, M., Kucerova, J., Horvathova, M., Divoky, V. Iron status in patients with pyruvate kinase deficiency: neonatal hyperferritinaemia associated with a novel frameshift deletion in the PKLR gene (p.arg518fs), and low hepcidin to ferritin ratios. Brit J Haematol May;165(4): IF 4.94 Příloha β: εethemoglobinémie (domácí publikace bez IF). εojzíková, R., Partschová, ε., Ko alková, P., Pospíšilová, D., Pokorná, P., εasárová, K., Divoký, V. První p ípad vrozené methemoglobinémie zp sobené sníţenou aktivitou cytochrom-b5-reduktázy v d sledku mutace v genu CYB5R3. Článek p ipraven k publikaci. Příloha γ : Talasemie (zahraniční publikace s IF) Divoka, M., Partschova, M., Kucerova, J., Mojzikova, R., Cermak, J., Pospisilova, D., Fabryova, V., Prochazkova, D., Indrak, K.,Divoky, V. εolekular Characterization of - Thalassemia in the Czech and Slovak populations: Mediterranean, Asian and Unique Mutations. Hemoglobin. Hemoglobin Jun; 40(3): IF Příloha 4: Talasemie (domácí publikace bez IF). Divoká ε., Partschová ε., Pospíšilová D., Orviská ε., δapčíková ů., Indrák K., Čermák J., Divoký V. Alfa-talasemie u δ5 českých rodin a γ7 rodin cizinc ţijících v České republice. P ehled literatury a molekulárně-genetická diagnostika. Trans Hemat Dnes. 22, 2016, No 3, p Příloha 5: Talasemie (zahraniční a domácí konference). Divoka, M., Partschova, M., Mojzikova, R., Piterkova, L., Horvathova, M., Cermak, J., Pospisilova, D., Indrak, K., Divoky, V. Molecular characterization of beta-thalassemia and 94
96 hemoglobin variants in the Czech and Slovak populations: an update. EHů, δondýn, UK, Haematol. Hematol J., vol 96 (Suppl. 2), p. 626 (1593). Publikovaný abstract. Divoka, M., Mojzikova, R., Piterkova, L., Pospisilova, P., Parstchova, M., Horvathova, M., Pospisilova, D., Striezencova Laluhova, Z., Cermak, J., Indrak, K., Divoky, V. Molecular characterization of hemoglobinopathies and red cell enzymopathies in the Czech and Slovak population: an update. ASH, San Diego, USA, Blood (ASH Annual Meeting Abstracts), 118(21), 2011, Online - publikovaný abstrakt. Divoká, ε., Partschová, ε., εojzíková, R., Piterková, δ., Horváthová, ε., Koláčková, ε., Kalandrová, E., Čermák, J., Pospíšilová, D., Fábryová, V., Indrák, K., Divoký, V. εolekulární charakterizace beta-talasemií a hemoglobinových variant v české a slovenské populaci. XXV. Olomoucké hematologické dny, Olomouc, ββ Trans Hemat Dnes, 17, 2011, str. 24. P ednáška. Divoká, ε., Koláčková, ε., δipert, J., Novosadová, ů., Orviská, ε., δapčíková, ů., δáníková, δ., Partschová, ε., Pospíšilová, D., Divoký, V., Indrák, K. Hemoglobinopatie v české a slovenské populaci. XXVII. Olomoucké hematologické dny s mezinárodní účastí. 1β. - 1δ. 5. β01γ, Olomouc. Sborník abstrakt, str. Ř5-86 (P43/2401). Poster. Příloha 6: εethemoglobinémie (domácí konference). Partschová, ε., εojzíková, R., Pospíšilová, P., Pospíšilová, D., εasárová, K., Divoký, V. εethemoglobinémie zp sobená sníţenou aktivitou cytochrom-b5-reduktázy: jedna nová mutace CYB5Rγ genu. XXVI. Olomoucké hematologické dny, Olomouc, βδ Sborník abstrakt, str. 71. Poster. Příloha 7: Hemochromatóza (zahraniční konference). Pospisilova, P., Mojzikova, R., Partschova, M., Zidova, Z., Horvathova, M., Pospisilova, D., Divoky, V. Hemolytic anemia with hyperferritinemia in Czech pyruvate kinase deficient patient a novel homozygous frameshift deletion in PKLR gene (p. R518fs) combined with TFR2 (p.r752h) and SLC40A1 (IVS1-24 C G) gene polymorphisms. 19th 95
97 ERCS meeting. Forteiland, IJmuiden, The Netherlands, Abstract book, p.47. P ednáška. 96
98
99 Příloha 1: Hemochromatóza (zahraniční publikace s IF). Mojzikova, R., Koralkova, P., Holub, D., Zidova, Z., Pospisilova, D., Cermak, J., Striezencova Laluhova, Z., Indrak, K., Sukova, M., Partschova, M., Kucerova, J., Horvathova, M., Divoky, V. Iron status in patients with pyruvate kinase deficiency: neonatal hyperferritinaemia associated with a novel frameshift deletion in the PKLR gene (p.arg518fs), and low hepcidin to ferritin ratios. Brit J Haematol May;165(4): IF 4.94
100 research paper Iron status in patients with pyruvate kinase deficiency: neonatal hyperferritinaemia associated with a novel frameshift deletion in the PKLR gene (p.arg518fs), and low hepcidin to ferritin ratios Renata Mojzikova, 1 * Pavla Koralkova, 1 * Dusan Holub, 2 Zuzana Zidova, 1 Dagmar Pospisilova, 3 Jaroslav Cermak, 4 Zuzana Striezencova Laluhova, 5 Karel Indrak, 6 Martina Sukova, 7 Martina Partschova, 1 Jana Kucerova, 1 Monika Horvathova 1 and Vladimir Divoky 1,6 1 Department of Biology, Faculty of Medicine and Dentistry, Palacky University, 2 Institute of Molecular and Translational Medicine, Faculty of Medicine and Dentistry, Palacky University, 3 Department of Paediatrics, University Hospital and Faculty of Medicine and Dentistry, Palacky University, Olomouc, 4 Institute of Haematology and Blood Transfusion, Prague, Czech Republic, 5 Children s Faculty Hospital with Policlinic, Bratislava, Slovak Republic, 6 Department of Haemato-Oncology, University Hospital and Faculty of Medicine and Dentistry, Palacky University, Olomouc, and 7 Department of Paediatric Haematology and Oncology, University Hospital Motol, Prague, Czech Republic Received 25 October 2013; accepted for publication 18 December 2013 Correspondence: Monika Horvathova Department of Biology, Faculty of Medicine and Dentistry Palacky University, Hnevotinska 3, Olomouc, Czech Republic. priwitzer@seznam.cz *These authors contributed equally to this work. Summary Pyruvate kinase (PK) deficiency is an iron-loading anaemia characterized by chronic haemolysis, ineffective erythropoiesis and a requirement for blood transfusion in most cases. We studied 11 patients from 10 unrelated families and found nine different disease-causing PKLR mutations. Two of these mutations - the point mutation c.878a>t (p.asp293val) and the frameshift deletion c.1553delg (p.(arg518leufs*12)) - have not been previously described in the literature. This frameshift deletion was associated with an unusually severe phenotype involving neonatal hyperferritinaemia that is not typical of PK deficiency. No disease-causing mutations in genes associated with haemochromatosis could be found. Inappropriately low levels of hepcidin with respect to iron loading were detected in all PK-deficient patients with increased ferritin, confirming the predominant effect of accelerated erythropoiesis on hepcidin production. Although the levels of a putative hepcidin suppressor, growth differentiation factor-15, were increased in PK-deficient patients, no negative correlation with hepcidin was found. This result indicates the existence of another as-yet unidentified erythroid regulator of hepcidin synthesis in PK deficiency. Keywords: red blood cell, pyruvate kinase deficiency, iron overload, hepcidin, ferritin. Erythrocyte pyruvate kinase (PK) deficiency, the most common cause of nonspherocytic haemolytic anaemia in central and northern Europe, is characterized by a variable degree of haemolysis and increased risk of iron overload. The mechanism of haemolysis is not clearly understood; however, the disruption of glycolysis leading to ATP depletion is one of the major contributing factors (Zanella et al, 2005). An appropriate diagnosis can be made only by measuring the erythrocyte PK enzyme activity and by direct mutation analysis of the gene encoding PK (PKLR). More than 220 mutations in the PKLR gene (1q22) have been identified worldwide since the first mutation was described in 1961 (Valentine et al, 1961). The disease is transmitted as a recessive trait, usually affecting only homozygotes or compound heterozygotes. The clinical manifestations vary from mild to severe anaemia requiring repeated transfusions. Rare cases of First published online 18 February 2014 doi: /bjh ª 2014 John Wiley & Sons Ltd British Journal of Haematology, 2014, 165,
101 Hepcidin in Pyruvate Kinase Deficiency neonatal death and non-immune hydrops foetalis have also been reported (Ferreira et al, 2000). Similar to patients with b-thalassaemia, PK-deficient patients may develop secondary iron overload. Although repeated blood transfusions were considered to be the major cause, iron accumulation also affects non-transfused PK-deficient patients (Zanella et al, 1993). The pathogenesis of iron overload appears to be multifactorial, involving chronic haemolysis, ineffective erythropoiesis, splenectomy and, eventually, coinheritance of hereditary haemochromatosis (Zanella et al, 2001). Importantly, the levels of hepcidin, the negative regulator of iron absorption in the gut and iron recycling from macrophages (Ganz, 2004), are reduced in patients with PK deficiency (Finkenstedt et al, 2008). Based on recent analyses, growth differentiation factor-15 (GDF15) is one of the candidate molecules proposed to suppress hepcidin production and enhance iron loading in the setting of ineffective erythropoiesis (Tanno & Miller, 2010; Tanno et al, 2010). The negative correlation between hepcidin and GDF15 was first reported in patients with b-thalassaemia (Tanno et al, 2007). Although GDF15 levels are increased in PK deficiency, the magnitude of GDF15 elevation is markedly lower than that in b-thalassaemia (Tanno et al, 2010). Here, we analysed PKLR mutations and iron status parameters in a cohort of 11 patients with PK deficiency. Our genetic data, together with calculations of the hepcidin/ferritin ratio, indicate the involvement of an as-yet unknown erythroid-derived hepcidin suppressor in PK deficiency. Design and methods Patients, haematological and biochemical analysis We investigated 11 patients with congenital nonspherocytic haemolytic anaemia from 10 unrelated Czech and Slovak families. The patient cohort consisted of five adults, three children and three infants. The Ethics Committee of Palacky University Hospital approved the collection and analysis of samples. Informed consent was obtained according to the Declaration of Helsinki. Red blood cell parameters were measured using a Sysmex XE-500 analyser (Sysmex Corp., Kobe, Japan). Serum parameters of iron metabolism, including iron, ferritin and transferrin saturation as well as bilirubin levels, were determined with standard biochemical methods. PK and hexokinase (HK) activities were measured in erythrocyte lysates that were free of leucocytes and platelets according to the methods recommended by the International Committee for Standardization in Haematology (Beutler et al, 1977). PK and HK activities were expressed as u/g haemoglobin (Hb). HK activity was included to estimate the contribution of young red blood cells to the total PK activity. The reference values for enzyme activities were established from 10 normal subjects. GDF15 serum concentrations were measured with an enzyme-linked immunosorbent assay (ELISA) kit for human GDF15 according to the manufacturer s instructions (R&D Systems, Minneapolis, MN, USA). The normal range for GDF15 was determined from eight healthy controls (four children and four adults). The serum erythropoietin (EPO) concentrations were measured by radioimmunoassay (Zadrazil et al, 2007). Hepcidin measurement The method for hepcidin measurement was established based on the protocol published by Li et al (2009). Briefly, each sample was prepared from 200 ll of human serum. Calibration standards were prepared from unmodified synthetic human hepcidin (DTHFPICIFCCGCCHRSKCGMCCKT) dissolved in rabbit serum. Hepcidin labelled with a stable isotope [ 13 C 9, 15 N 1 -Phe 4 ] (50 ng) was added to each sample as an internal standard. Both hepcidin variants were synthesized by Thermo Fisher Scientific (Ulm, Germany). The samples were extracted on an Oasis HLB lelution plate and separated by reverse-phase liquid chromatography using an UltiMate 3000 Nano LC System (Thermo Fisher Scientific, Sunnyvale, CA, USA) coupled to a QTRAP 5500 mass spectrometer (AB SCIEX, Framingham, MA, USA). Subsequent detection of hepcidin was performed by selected reaction monitoring method. The acquired data were processed using Analyst â software (AB SCIEX, version 1.5.1). The assay was validated by Intrinsic LifeSciences LLC (La Jolla, CA, USA), and the normal control range was determined in 18 healthy individuals (10 children and eight adults). Molecular analysis Genomic DNA was isolated from peripheral blood drawn in EDTA using a QIAamp DNA Blood Maxi Kit (Qiagen, Valencia, CA, USA). The exons of the following were amplified by polymerase chain reaction (PCR): PKLR (accession: NM_ ) (Lenzner et al, 1997); genes known to be responsible for hereditary haemochromatosis (Camaschella & Poggiali, 2011), including HFE (accession: NM_ ), HFE2 (haemojuvelin) (accession: NM_ ), HAMP (hepcidin) (accession: NM_ ), TFR2 (transferrin receptor 2) (accession: NM_ ) and SLC40A1 (ferroportin 1) (accession: NM_ ); and a gene encoding the light chain of ferritin (FTL) (accession: NM_ ). The iron response element (IRE) regions of the 5 0 -UTR of SLC40A1 and FTL were also amplified. PCR primers and conditions are available upon request. The PCR products were purified with a QIAquick kit (Qiagen) and sequenced using the BigDye terminator kit (Applied Biosystems, Foster City, CA, USA). Sequence analysis was performed using the ABI Prism 310 Genetic Analyser (Applied Biosystems) with software provided by the manufacturer. Statistical methods Student s t-test was used to determine the statistical difference between the patients and controls using Origin 6.1 ª 2014 John Wiley & Sons Ltd 557 British Journal of Haematology, 2014, 165,
102 R. Mojzikova et al software (OriginLab Corporation, Northampton, MA, USA). The Spearman coefficient for correlation analyses was calculated using Statistica 10 software (StatSoft, Inc., Tulsa, OK, USA). P values <005 were considered statistically significant. Results Patients characteristics and PK activity The clinical findings of nine unrelated patients and two siblings obtained at the time of diagnosis are summarized in Table I. All patients suffered from mild to severe haemolytic anaemia (with Hb levels ranging from 65 to 121 g/l) and hyperbilirubinaemia (33 to 172 lmol/l). All but one of these patients had increased reticulocyte counts (48 to 805%). Neonatal jaundice was reported in most of the patients. Seven patients were transfusion-dependent; three were concomitantly treated with chelators and two underwent splenectomy. Two paediatric patients received transfusions during infancy, one adult patient was on intermittent transfusion therapy and only one patient had never received a transfusion in his lifetime. In 10 patients, erythrocyte PK activity was below the normal reference values ( u/g Hb) (Table II). In one case (Patient 7), the activity was false normal (658 u/g Hb) due to substantial reticulocytosis (805%) and PK deficiency was therefore established based on low PK/HK activity ratio (16; normal range 36 63) (Table II). Reduced PK activity compared with the controls was also detected in the available healthy parents, confirming a congenital defect in the child. All parents tested were later shown to be heterozygous carriers of specific PKLR mutations (Table II). Mutational analysis of the PKLR gene Direct sequence analysis of the PKLR gene in the patient cohort revealed nine different mutations in either the homozygous state (four patients) or the compound heterozygous state (seven patients) (Table II). Seven of the mutations are known; five of these (c.1594c>t, c.1529g>a, c.1493g>a, c.1456c>t, and c.994g>a) have previously been identified in Czech and Slovak populations (Lenzner et al, 1997). The c.823g>a substitution and the insertion-deletion (c.347_350delinsaacattg) were identified in these populations for the first time. The clinical picture and the severity of the disease in our patients bearing the above-mentioned PK mutations were comparable to the phenotype previously reported for patients with identical mutations (Baronciani et al, 1995; Zanella et al, 1997, 2005, 2007). The c.878a>t and c.1553delg mutations are novel and have not been previously described in the literature. The maternally inherited c.878a>t missense mutation was detected in a heterozygous state with the previously reported, paternally inherited c.1529g>a substitution (Fig 1A, Table II), which is the most common PKLR mutation in the northern and central European population (Zanella et al, 2005). The c.878a>t mutation involves the alteration of positively charged aspartic acid to hydrophobic valine on the Table I. Clinical and haematological data of the patients. Patient Sex Age Neonatal jaundice ExTx Spleen Hb, g/l Ret,% [05 30] Bilirubin, lmol/l [0 23] Ferritin, lg/l [6 200]* TfS [ ] Hepcidin, lg/l [ ] GDF15, ng/l [ ] EPO, iu/l [43 29] 1 M 9 years Yes Yes Out n.d n.d 2 M 6 months Yes Yes In months n.d F 7 years Yes Yes In M 10 months Yes Yes In n.d M 7 years Yes Yes In n.d 6 F 42 years No Yes In F 22 years Yes Yes Out F 31 years Yes Yes In M 3 months Yes Yes In n.d. n.d n.d 10 F 25 years Yes Yes In n.d 11 M 50 years No No In Patients 3 and 4 are siblings; ExTx: exchange transfusion; TfS: transferrin saturation; GDF15: growth differentiation factor-15; EPO (serum erythropoietin); n.d.: not determined. *normal range for ferritin levels, women: <200, men: <300, children: <70 lg/l (Brugnara, 2009). The control range for GDF15 was determined from eight healthy controls (four children and four adults). Dependent on blood transfusion during the first year of life due to severe anaemia. Intermittent ExTx. Patients on chelation therapy. Two sets of data are shown for Patient 2; the first set represents values at the time of diagnosis, the second corresponds to values after 6 months of chelation therapy. 558 ª 2014 John Wiley & Sons Ltd British Journal of Haematology, 2014, 165,

103 Hepcidin in Pyruvate Kinase Deficiency Table II. Biochemical and molecular data of the patients and their family members. Patient PK activity, u/g Hb PK/HK PKLR mutation Amino acid alteration c.[347_350delinsaacattg]; [347_350delinsAACATTG] p.[arg116_leu117delinsglnhiscys]; [Arg116_Leu117delinsGlnHisCys] 1 (mother) c.[347_350delinsaacattg];[=] p.[arg116_leu117delinsglnhiscys];[=] 1 (father) c.[347_350delinsaacattg];[=] p.[arg116_leu117delinsglnhiscys];[=] c.[1553delg];[1553delg] p.[arg518leufs*12];[arg518leufs*12] 2 (mother) c.[1553delg];[=] p.[arg518leufs*12];[=] c.[1529g>a];[1594c>t] p.[arg510gln];[arg532trp] c.[1529g>a];[1594c>t] p.[arg510gln];[arg532trp] 4 (mother) c.[1529g>a];[=] p.[arg510gln];[=] 4 (father) c.[1594c>t];[=] p.[arg532trp];[=] c.[1493g>a];[1529g>a] p.[arg498his];[arg510gln] c.[1456c>t];[1594c>t] p.[arg486trp];[arg532trp] c.[1594c>t];[1594c>t] p.[arg532trp];[(arg532trp)] c.[878a>t];[1529g>a] p.[asp293val];[arg510gln] 8 (father) c.[1529g>a];[=] p.[arg510gln];[=] 8 (mother) c.[878a>t];[=] p.[asp293val];[=] c.[823g>a];[1594c>t] p.[gly275arg];[arg532trp] 9 (father) c.[823g>a];[=] p.[gly275arg];[=] 9 (mother) c.[1594c>t];[=] p.[arg532trp];[=] c.[1529g>a];[1529g>a] p.[arg510gln];[(arg510gln)] c.[994g>a];[1456c>t] p.[gly332ser];[arg532trp] PK: pyruvate kinase (normal activity range: u/g Hb); PK/HK: pyruvate kinase and hexokinase activity ratio (normal range: 36 63). The normal activity range was determined in 10 healthy controls. (A), exon 7, exon 11 (B), exon 11 C A C T T A T G C CN A G G A G T C T T C Patient 8 Wild type C A C T T A T G C C G A G G A G T C T T C Mother Patient 2 C A C T T A TG C C N A G G A G T C T T C Father Mother Fig 1. Detection of two novel PKLR mutations by sequencing amplified DNA. (A) In the family of Patient 8, the propositus carried the newly described c.878a>t mutation in exon 7 and the c.1529g>a mutation in exon 11; the mother was heterozygous for the c.878a>t mutation, and the father was heterozygous for the c.1529g>a mutation. (B) In the family of Patient 2, the propositus was homozygous for the second newly described mutation in the PKLR gene, a single nucleotide deletion (c.1553delg) in exon 11 leading to a frameshift (p.(arg518leufs*12)) and a premature stop codon. One allele was inherited from his heterozygous mother; his father was unavailable for the analyses. ª 2014 John Wiley & Sons Ltd 559 British Journal of Haematology, 2014, 165,
104 R. Mojzikova et al surface of the A domain of PK. This alteration might increase the hydrophobicity of PK, which is thermodynamically less favourable for protein folding. PK activity was markedly reduced to 156 u/g Hb in an affected patient (Patient 8) who presented with a mild phenotype (Hb 95 g/l, reticulocytes 66%, and bilirubin 34 lmol/l; Table I). The novel PKLR frameshift deletion associated with atypical severe phenotype involving neonatal hyperferritinaemia Patient 2 carried the second previously unidentified frameshift deletion, c.1553delg (p.(arg518leufs*12), short p.arg518fs), in a homozygous state; the mother is a heterozygous carrier of this deletion (Fig 1B, Table II). Because the father is unknown and thus unavailable for analyses, a possible hemizygous genomic deletion (loss of heterozygosity) was excluded by quantitative PCR-based copy number analysis (Supplementary methods; data not shown). The mutation causes premature termination of translation at codon 529, with predicted formation of a truncated protein lacking 46 C-terminal amino acids including the critical activator binding site Arg532 (Valentini et al, 2002). The activity of PK was reduced in both the patient (163 u/g Hb) and his mother (257 u/g Hb). The patient s clinical picture was very severe from birth, with a need for repeated blood transfusions. The patient exhibited profound anaemia (Hb, 77 g/l), with signs of hypoxia, neonatal jaundice (bilirubin, 90 lmol/l), thrombocytopenia, prominent hepatosplenomegaly and dramatic hyperferritinaemia (in the range of lg/l), and transferrin saturation reaching almost 100%. Elevated levels of liver enzymes (not shown) suggested possible liver damage, which led us to perform liver biopsy. Massive iron deposits in both hepatocytes and Kupffer cells corresponding to grade IV haemosiderosis were detected. Taken together, these findings suggested the presence of another co-inherited defect in the child aside from PK deficiency. Sideroblastic anaemia was ruled out by the absence of sideroblasts in the bone marrow. The nucleotide sequence of the IRE region of the FTL gene, which encodes L-ferritin, was not mutated; thus, a possible association with hereditary hyperferritinaemia cataract syndrome (HHCS) was excluded (Brooks et al, 2002). Sequence analysis of all known genes that cause primary haemochromatosis (HFE, HFE2, HAMP, SLC40A1 and TFR2) (Camaschella & Poggiali, 2011) did not reveal any causative mutations. Only five different previously described SNP variations were identified in TFR2 (c.2255g>a (p.arg752his) - rs ), SLC40A1 (c.44-24g>c - rs , c.-330cgg[8] - rs , c.663t>c (p.val221 = ) - rs ) and FTL (c.163t>c (p.leu55 = )) (Table SI). Of these polymorphisms, only rs :g>c was previously shown to be associated with the clinical aggressiveness (liver damage) of hereditary haemochromatosis 1 caused by the p.cys282tyr HFE mutation; however, this SNP had no effect on ferritin levels in the control group, and it was therefore concluded that the negative effect of the rs :c allele was restricted to pathological conditions (Altes et al, 2009). Because nine of our remaining PK-deficient patients were also homozygous for the rs :c allele (Table SI), this polymorphism probably does not contribute to the severe phenotype of this patient and cannot explain his iron overload. Recently, at an age of two and a half years, the patient s ferritin levels dramatically decreased (to 145 lg/l), which may be a result of combined chelation therapy lasting 6 months and a prolonged interval between transfusions. Based on these new findings, we conclude that the severe neonatal phenotype of this patient was caused by the PKLR mutation itself. Iron status in PK-deficient patients One of the main health complications in PK deficiency is secondary iron overload with multifactorial pathogenesis (Zanella et al, 1993, 2001). In our cohort, nine patients had elevated serum ferritin (median, 724 lg/l), including the patient with no transfusion history (Patient 11). Of nine available cases, five showed a concomitant increase in transferrin saturation (from 050 to 097) (Table I). Firstly, we assessed a possible effect of splenectomy on iron loading. One of our two splenectomized patients (Patient 7) showed a progressive increase in ferritin levels after spleen removal. Nevertheless, sustained haemolysis was observed in this patient after spleen surgery, which probably contributed to a further increase in her ferritin levels. On the other hand, no significant difference in ferritin was recorded for the second splenectomized patient (Patient 1); splenectomy improved this patient s anaemia, nevertheless his presplenectomy ferritin levels were already dramatically elevated (2213 lg/l). This patient is a child; thus, the contribution of spleen removal to iron loading may become more apparent over time. We next searched for the presence of known HFE mutations (p.his63asp, p.cys282tyr), which have been proposed as a condition that may predispose a patient to increased iron absorption (Arruda et al, 2000; Zanella et al, 2001). Abnormal HFE genotypes were identified in four of our patients (Table SI). One of these patients (Patient 3), who was a p.[his63asp];[cys282tyr] HFE compound heterozygote, was the only paediatric patient from the group with normal ferritin levels. Hepcidin levels in PK deficiency To address the role of hepcidin in the pathogenesis of iron overload in our patient cohort, we established a proteomicsbased method for hepcidin measurement. As expected, the levels of hepcidin were reduced in PK-deficient patients (median, 158 lg/l) (Table I) compared with healthy agematched controls (median, 276 lg/l) (Table I), reflecting the impact of ineffective erythropoiesis in PK deficiency. The difference, however, was not statistically significant. We 560 ª 2014 John Wiley & Sons Ltd British Journal of Haematology, 2014, 165,
105 Hepcidin in Pyruvate Kinase Deficiency therefore calculated the hepcidin/ferritin ratio which represents a more accurate estimation of proper hepcidin production with respect to iron loading, and found it to be very low in our PK-deficient patients (median, 006) compared with a healthy age-matched group of controls (median, 035) (Fig 2). Even the patient with the highest hepcidin level (Patient 2) showed a dramatically reduced ratio, which was detected in repeated measurements (ranging from 001 to 004) and eventually increased to 011 as a response to 6 months of chelation therapy and improved anaemia (Hb, 98 g/l) (Table I and Fig 2). The direct effect of chelation therapy on hepcidin levels and hepcidin/ferritin ratio is difficult to address in our cohort, given the complexity of positive and negative signals regulating hepcidin, the small number of patients receiving chelation therapy, and the lack of hepcidin pre-chelation values for remaining patients. Candidate erythroid signals regulating hepcidin To clarify the involvement of erythropoiesis in iron metabolism in PK deficiency, serum levels of GDF15 and EPO, two markers of erythropoietic activity known to be associated with hepcidin production, were evaluated. The levels of GDF15, a putative negative regulator of hepcidin production in the setting of ineffective erythropoiesis (Tanno & Miller, 2010), were determined using a commercial enzyme-linked immunosorbent assay. PK-deficient patients showed elevated GDF15 compared with healthy age-matched controls Hepcidin/ferritin ratio 2 5 < Controls PKD patients Fig 2. Hepcidin/ferritin ratio in PK-deficient patients. The hepcidin/ ferritin ratio was significantly lower in PK-deficient patients compared to healthy controls (P < 005). The red dots in the graph indicate the hepcidin/ferritin ratio calculated from five repeated measurements (15 months, 95 months, 10 months, 205 months, and 33 months) for Patient 2 with a p.(arg518leufs*12) mutation in the PKLR gene, which is associated with hyperferritinaemia occurring from the neonatal period through to infancy (2 years of age). Only two hepcidin/ferritin ratios (calculated from those hepcidin and ferritin values shown in Table I) were included into the statistical analysis for this patient. The thick horizontal lines represent the medians for each group. PKD, pyruvate kinase-deficient. (median, 9055 lg/l and 223 lg/l, respectively) (Table I), confirming previously reported results (Zanella et al, 2005). Importantly, no correlation between GDF15 and hepcidin or GDF15 and hepcidin/ferritin ratio was observed. In addition, all available patients from our cohort showed increased levels of EPO (median, 1001 iu/l) (Table I), probably reflecting tissue hypoxia. Also, EPO levels did not correlate with hepcidin or hepcidin/ferritin ratio. Lastly, as inflammatory processes are known to independently influence hepcidin levels (Ganz, 2004), it is important to note that none of our patients had signs of inflammation at the time of the analyses. In concordance, a substantial increase in hepcidin (from 907 lg/l to 386 lg/l) was observed in one of our patients (Patient 2) during bacterial infection. Discussion In this report we characterized PKLR mutations and systemic iron metabolism in eleven patients with PK deficiency from ten unrelated families. Nine different disease-causing PKLR mutations were identified; two of these mutations - the point mutation c.a878t (p.d293v) and the frameshift deletion c.1553delg (p.r518fs) - were novel. The patient affected by the frameshift deletion presented with an unusually severe phenotype involving neonatal hyperferritinaemia, which eventually ameliorated by two and a half years of age, probably in response to combined chelation therapy and a prolonged interval between transfusions. PK-deficient patients usually develop secondary iron overload over time as a consequence of chronic haemolysis, ineffective erythropoiesis and transfusion therapy; splenectomy and inheritance of HFE mutations have been proposed as additional risk factors (Zanella et al, 2001). Consistently, all our transfusion-dependent patients presented iron overload; one patient has developed hyperferritinaemia independently of blood transfusions. Similar findings, iron overload independent of blood transfusions in PK deficiency, were previously reported by Zanella et al (1993). Although splenectomy reduces the symptoms of haemolysis, it is also known to be a risk factor for iron loading. Due to a low number of splenectomized patients in our cohort, however, the involvement of spleen surgery cannot be adequately addressed. The contribution of co-inherited HFE mutations to excessive iron accumulation is also difficult to assess, especially because three of the HFE mutant patients were children and HFE-related haemochromatosis is an adultonset disease (Pietrangelo, 2010). Moreover, one paediatric patient with abnormal HFE genotype did not have any signs of iron overload. Based on these observations we can conclude that the HFE genotype does not play a major role in determining iron loading in PK-deficient paediatric patients; however, it may be important to follow up these patients due to a potential higher risk of iron overload in adulthood (Zanella et al, 2001). ª 2014 John Wiley & Sons Ltd 561 British Journal of Haematology, 2014, 165,
106 R. Mojzikova et al Analysis of factors regulating iron homeostasis revealed that hepcidin levels were relatively low for the degree of iron loading in all PK-deficient patients with increased ferritin. Similar results were published for b-thalassaemia intermedia (Kearney et al, 2007; Origa et al, 2007), which clearly confirm the suppression of hepcidin by an erythroid signal that overrides iron loading-induced signalling. Although the levels of the putative hepcidin suppressor, GDF15, were increased in our patient cohort, the levels were considerably lower than those reported for b-thalassaemia (Tanno et al, 2010). No correlation between GDF15 and hepcidin or hepcidin/ferritin ratio indicates that GDF15 can only be used as a marker of accelerated ineffective erythropoiesis in PK deficiency (Tanno & Miller, 2010; Tanno et al, 2010). Besides elevated GDF15 in the patients serum, we also found increased levels of EPO (Ashby et al, 2010; Ganz, 2011) that again did not correlate with hepcidin. This evidence further suggests that EPO by itself is an indirect suppressor of hepcidin as mice with disrupted erythropoiesis are not able to attenuate hepcidin synthesis in response to EPO administration (Pak et al, 2006; Vokurka et al, 2006). In addition to GDF15 and EPO, a suppressive effect of twisted gastrulation protein homolog 1 (TWSG1) (Tanno et al, 2009) and hypoxia (Yoon et al, 2006; Ganz, 2011) should be considered with respect to the regulation of hepcidin production. Nevertheless, the role of TWSG1 has not been clearly confirmed so far (Tanno et al, 2010). Although hypoxia inducible factor (HIF) signalling was reported to directly and also indirectly (through EPO) downregulate hepcidin (Gordeuk et al, 2011), the exact signalling remains elusive. Altogether, our findings indicate that the main erythroid-derived regulator of hepcidin synthesis in PK deficiency remains to be identified. In conclusion, we have shown that the hepcidin/ferritin ratio is consistently low in PK deficiency, which reflects relatively low hepcidin levels, even in PK deficiency associated with hyperferritinaemia. Acknowledgements This work was supported by grant NT/11208 (Ministry of Health, Czech Republic) and partially supported by grant LF_2013_010 (Internal Grant Agency of Palacky University). DH was partially supported by CZ.1.05/2.1.00/ (Ministry of Education, Youth and Sports, Czech Republic), MS was supported by NT/13587 (Ministry of Health, Czech Republic) and MH and VD were partially supported by P305/11/1745 (Czech Science Foundation). We thank Prof. Thomas Ganz (UCLA) for the validation of our HPLC-MS-based hepcidin measurements, Prof. Tomas Adam (Palacky University) for some biochemical measurements, and Lenka Radova (Masaryk University in Brno, Czech Republic) for statistical analyses. Author contributions RM and PK designed the study, performed enzyme assays and some molecular analyses, collected and analysed data and contributed to manuscript writing. DH established the hepcidin assay. ZZ determined GDF15 levels. DP, JC, ZSL, KI and MS treated the patients, collected patient material and provided clinical information. MP and JK participated in molecular analyses. MH contributed to the design of the study, interpretation of the results and wrote the manuscript. VD participated in the design of the study, interpretation of the results and revision/editing of the manuscript. Conflict of interest The authors declare no competing financial interests. Supporting Information Additional Supporting Information may be found in the online version of this article: Methods S1. qpcr-based copy number analysis. Table SI. HFE, TFR2 and SLC40A1 SNPs. References Altes, A., Bach, V., Ruiz, A., Esteve, A., Remacha, A.F., Sarda, M.P., Felez, J. & Baiget, M. (2009) Does the SLC40A1 gene modify HFE-related haemochromatosis phenotypes? Annals of Hematology, 88, Arruda, V.R., Agostinho, M.F., Cancado, R., Costa, F.F. & Saad, S.T. (2000) beta-thalassemia trait might increase the severity of hemochromatosis in subjects with the C282Y mutation in the HFE gene. American Journal of Hematology, 63, 230. Ashby, D.R., Gale, D.P., Busbridge, M., Murphy, K.G., Duncan, N.D., Cairns, T.D., Taube, D.H., Bloom, S.R., Tam, F.W., Chapman, R., Maxwell, P.H. & Choi, P. (2010) Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica, 95, Baronciani, L., Magalh~aes, I.Q., Mahoney, D.H. Jr, Westwood, B., Adekile, A.D., Lappin, T.R. & Beutler, E. (1995) Study of the molecular defects in pyruvate kinase deficient patients affected by nonspherocytic hemolytic anemia. Blood Cells, Molecules & Diseases, 21, Beutler, E., Blume, K.G., Kaplan, J.C., Lӧhr, G.W., Ramot, B. & Valentine, W.N. (1977) International committee for standardization in haematology: recommended methods for red-cell enzyme analysis. British Journal of Haematology, 35, Brooks, D.G., Manova-Todorova, K., Farmer, J., Lobmayer, L., Wilson, R.B., Eagle, R.C. Jr, St Pierre, T.G. & Stambolian, D. (2002) Ferritin crystal cataracts in hereditary hyperferritinemia cataract syndrome. Investigative Ophtalmology & Visual Science, 43, Brugnara, C.. (2009) Reference values in infancy and childhood. In: Nathan and Oski s Hematology of Infancy and Childhood. (eds. by S.H. Orkin, D.E. Fisher, A.T. Look, S.E. Lux, D. Ginsburg & D.G. Nathan), 7th edn, pp Saunders elsevier, Philadelphia. Camaschella, C. & Poggiali, E. (2011) Inherited disorders of iron metabolism. Current Opinion in Pediatrics, 23, Ferreira, P., Morais, L., Costa, R., Resende, C., Dias, C.P., Araujo, F., Costa, E., Barbot, J. & Vilarinho, A. (2000) Hydrops fetalis associated with erythrocyte pyruvate kinase deficiency. European Journal of Pediatrics, 159, ª 2014 John Wiley & Sons Ltd British Journal of Haematology, 2014, 165,
107 Hepcidin in Pyruvate Kinase Deficiency Finkenstedt, A., Bianchi, P., Theurl, I., Vogel, W., Witcher, D.R., Wroblewski, V.J., Murphy, A.T., Zanella, A. & Zoller, H. (2008) Regulation of iron metabolism through GDF15 and hepcidin in pyruvate kinase deficiency. Britsh Journal of Haematology, 144, Ganz, T. (2004) Hepcidin in iron metabolism. Current Opinion in Hematology, 11, Ganz, T. (2011) Hepcidin and iron regulation, 10 years later. Blood, 117, Gordeuk, V.R., Miasnikova, G.Y., Sergueeva, A.I., Niu, X., Nouraie, M., Okhotin, D.J., Polyakova, L.A., Ammosova, T., Nekhai, S., Ganz, T. & Prchal, J.T. (2011) Chuvash polycythemia VHLR200W mutation is associated with downregulation of hepcidin expression. Blood, 118, Kearney, S.L., Nemeth, E., Neufeld, E.J., Thapa, D., Ganz, T., Weinstein, D.A. & Cunningham, M.J. (2007) Urinary hepcidin in congenital chronic anemias. Pediatric Blood & Cancer, 48, Lenzner, C., N urnberg, P., Jaobasch, G., Gerth, C. & Thiele, B.J. (1997) Molecular analysis of 29 pyruvate kinase-deficient patients from central Europe with hereditary hemolytic anemia. Blood, 89, Li, H., Rose, M.J., Tran, L., Zhang, J., Miranda, L.P., James, C.A. & Sasu, B.J. (2009) Development of a method for the sensitive and quatitative determination of hepcidin in human serum using LC-MS/MS. Journal of Pharmacological and Toxicological Methods, 59, Origa, R., Galanello, R., Ganz, T., Giagu, N., Maccioni, L., Faa, G. & Nemeth, E. (2007) Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica, 92, Pak, M., Lopez, M.A., Gabayan, V., Ganz, T. & Rivera, S. (2006) Suppression of hepcidin during anemia requires erythropoietic activity. Blood, 108, Pietrangelo, A. (2010) Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology, 139, , 408.e1-2. Tanno, T. & Miller, J.L. (2010) Iron loading and overloading due to ineffective erythropoiesis. Advances in Hematology, doi: /2010/ Tanno, T., Bhanu, N.V., Oneal, P.A., Goh, S.H., Staker, P., Lee, Y.T., Moroney, J.W., Reed, C.H., Lubn, N.L., Wang, R.H., Eling, T.E., Childs, R., Ganz, T., Leitman, S.F., Fucharoen, S. & Miller, J.L. (2007) High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nature Medicine, 13, Tanno, T., Porayette, P., Sripichai, O., Noh, S.J., Byrnes, C., Bhupatiraju, A., Lee, Y.T., Goodnough, J.B., Harandi, O., Ganz, T., Paulson, R.F. & Miller, J.L. (2009) Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood, 114, Tanno, T., Noel, P. & Miller, J.L. (2010) Growth differentiation factor 15 in erythroid health and disease. Current Opinion in Haematology, 17, Valentine, W.N., Tanaka, K.R. & Miwa, S. (1961) A specific erythrocyte glycolytic enzyme defect (pyruvate kinase) in three subjects with congenital non-spherocytic hemolytic anemia. Transactions of the Association of American Physicians, 74, Valentini, G., Chiarelli, L.R., Fortin, R., Dolzan, M., Galizzi, A., Abraham, D.J., Wang, C., Bianchi, P., Zanella, A. & Mattevi, A. (2002) Structure and function of human erythrocyte pyruvate kinase. Molecular basis of nonspherocytic hemolytic anemia. The Journal of Biological Chemistry, 277, Vokurka, M., Krijt, J., Sulc, K. & Necas, E. (2006) Hepcidin mrna levels in mouse liver respond to inhibition of erythropoiesis. Physiological Research, 55, Yoon, D., Pastore, Y.D., Divoky, V., Liu, E., Mlodnicka, A.E., Rainey, K., Ponka, P., Semenza, G.L., Schumacher, A. & Prchal, J.T. (2006) Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. The Journal of Biological Chemistry, 281, Zadrazil, J., Horak, P., Horcicka, V., Zahalkova, J., Strebl, P. & Hruby, M. (2007) Endogenous erythropoietin levels and anemia in long-term renal transplant recipients. Kidney and Blood Pressure Research, 30, Zanella, A., Berzuini, A., Colombo, M.B., Guffanti, A., Lecchi, L., Poli, F., Cappellini, M.D. & Barosi, G. (1993) Iron status in red cell pyruvate kinase deficiency: study of Italian cases. British Journal of Haematology, 83, Zanella, A., Bianchi, P., Baronciani, L., Zappa, M., Bredi, E., Vercellati, C., Alfinito, F., Pelissero, G. & Sirchia, G. (1997) Molecular characterization of PK-LR gene in pyruvate kinase deficient patients. Blood, 89, Zanella, A., Bianachi, P., Iurlo, A., Boschetti, C., Taioli, E., Verellati, C., Zappa, M., Fermo, E., Tavazzi, D. & Sampietro, M. (2001) Iron status and HFE genotype in erythrocyte pyruvate kinase deficiency: study of Italian cases. Blood Cells Molecules and Diseases, 27, Zanella, A., Fermo, E., Bianchi, P. & Valentini, G. (2005) Red cell pyruvate kinase deficiency: molecular and clinical aspects. British Journal of Haematology, 130, Zanella, A., Fermo, E., Bianchi, P., Chiarelli, L.R. & Valentini, G. (2007) Pyruvate kinase deficiency: the genotype-phenotype association. Blood Reviews, 21, ª 2014 John Wiley & Sons Ltd 563 British Journal of Haematology, 2014, 165,
108 Supplementary Method. Copy number of PKLR exon 10 was assessed by SYBR Green I based absolute quantification, using standard curve method. Concentration values of PKLR were normalized to RPPH1 housekeeping gene that is commonly used as a copy number variation (CNV) reference, as it s an autosomal gene with no reported CNV in population. The reaction was run in triplicates. The copy number value of Patient 2 was compared to six non-pk-deficient patient samples. The primer sequences are available upon request.
109 Table SI. HFE, TFR2 and SLC40A1 SNPs. Patient HFE mutation TFR2 p.arg752his SLC40A1 CGG(7_8) SLC40A1 c.44-24g>c 1 wt wt CGG[7];[8] [C];[C] 1 (mother) wt wt CGG[8];[8] [C];[C] 1 (father) wt wt CGG[7];[7] [C];[C] 2 wt [Arg752His];[=] CGG[8];[8] [C];[C] 2 (mother) wt wt CGG[8];[8] [C];[C] 3 [His63Asp];[Cys282Tyr] wt CGG[8];[8] [C];[C] 4 [His63Asp];[=] wt CGG[8];[8] [C];[C] 3,4 (mother) [Cys282Tyr];[=] wt CGG[8];[8] [C];[C] 3,4 (father) [His63Asp];[=] wt CGG[7];[8] [C];[C] 5 [His63Asp];[=] wt CGG[8];[8] [C];[C] 6 [His63Asp];[(His63Asp)] wt CGG[8];[8] [C];[C] 7 wt wt CGG[7];[8] [C];[C] 8 wt wt CGG[7];[8] [C];[C] 8 (mother) wt wt CGG[7];[7] [C];[G] 8 (father) wt wt CGG[7];[8] [C];[C] 9 wt wt CGG[7];[8] [C];[G] 9 (mother) wt wt CGG[8];[8] [C];[C] 9 (father) wt wt CGG[7];[8] [C];[G] 10 wt wt CGG[8];[8] [C];[C] 11 wt wt CGG[8];[8] [C];[C] Rem.: HFE screened for p.cys282tyr, p.his63asp, p.ser65cys, and p.val256ile; TFR2 for p.arg752his and SLC40A1 for microsatellite polymorphism c.-330cgg(7_8) and c.44-24g>c. CGG[7];[8] in SLC40A1column is short for c.-330cgg[7];[8]. [C];[C] and [C];[G] in SLC40A1column is short for c.[44-24g>c];[ 44-24G>C] and c.[44-24g>c];[=], respectively.
110 Příloha 2: Methemoglobinémie (domácí publikace bez IF). Mojzíková, R., Partschová, M., Kořalková, P., Pospíšilová, D., Pokorná, P., Masárová, K., Divoký, V. První případ vrozené methemoglobinémie způsobené sníženou aktivitou cytochrom-b5-reduktázy v důsledku mutace v genu CYB5R3. Článek připraven k publikaci.
111 Prv í případ vroze é ethe oglo i é ie typu I v české popula i způso e é s íže ou aktivitou cytochrom-b5-reduktasy v důsledku dvojité mutace v genu CYB5R3 Mojzíková R 1, Parts hová M 1, Kořalková P 1, Pospíšilová D 2, Pokor á P 3, Masárová K 4, Divoký V 1,5 1 Ústav iologie, Lékařská fakulta U iverzit Pala kého v Olo ou i 2 Dětská kli ika, Fakult í e o i e a Lékařská fakulta U iverzit Pala kého v Olo ou i 3 Všeo e á fakult í e o i e v Praze 4 Kli ika he atológie a tra sfuziológie, Fakult á e o i a s poliklinikou Bratislava 5 Hemato-o kologi ká kli ika, Fakult í e o i e a Lékařská fakulta U iverzit Pala kého v Olomouci
112 SOUHRN E z ová ethe oglo i é ie je autoso ál ě re esiv í o e o ě í způso e é defi ite enzymu cytochrom-b5-reduktas, který se v er tro te h podílí a zpět é reduk i methemoglobinu. V sou oru pa ie tů lo u jed oho z i h prokázá o, že se jed á o dvojitého heteroz gota pro mutaci v genu CYB5R3 kódují í te to e z, vedou í v tomto případě k jeho 18 % z tkové aktivitě. Jed á se o již popsa ou uta i. G A (p.val253met) a novou mutaci c.536c A (p.ala179glu). Fe ot pově se jed á o ethe oglo i é ii t pu I s methemoglobinem v roz ezí až %. U další h tře h pa ie tů s ír ou resp. as pto ati kou ethe oglo i é ií ethe oglo i -11 % ve 2 případe h a terapii k s. askor ovou la deteková a heteroz got í uta e v genu CYB5R3 způso ují í 5% s íže í e z ové aktivit. Jed á se o epopsa ou uta i p.met Val, vzá ý pol orfis us p.arg His a uta i p.val Met. Protože se jed á o heteroz got í uta e elze kauzalitu o ou prv í h uta í ez podro ější h fu kč í h studií jed oz ač ě sta ovit. U třetí uta e lo popsá o, že v ho oz got í for ě vede k ethe oglo i é ií typu I. Jed á se o prv í případ zá h tu e z ové ethe oglo i é ie v české a slove ské populaci. Klíčová slova: ethe oglo i é ie, defi it to hro -b5-reduktasy SUMMARY Congenital methemoglobinemia due to cytochrome b5 reductase deficiency is an autosomal recessive disorder. The major role of this enzyme in the erythrocytes is the methemoglobin reduction. In a cohort of four patients, one was diagnosed to be double heterozygote for mutation in CYB5R3 gene encoding this enzyme, which leads to 18 % of its residual activity. The identified mutations were mutation c.757g A (p.val253met) and a new mutation c.536c A (p.ala179glu). The patient displays Type-I phenotype with methemoglobin levels ranged from 9 to 22 %. Three other patients with methemoglobinemia (methemoglobin levels ranged from 7 to 11 % two patients are on ascorbic acid therapy) were confirmed to be single heterozygote for CYB5R3 gene mutation with 75% decrease of enzyme activity. Three different mutations
113 were identified - one novel mutation p.met134val, p.arg297his (rare polymorfism) and p.val106met. All these mutations were found in the heterozygote form, thus the causality of the mutation p.mt134val and p.arg297his could not be confirmed without further functional studies. Homozygous mutation p.val106met leads to type I methemoglobinemia as was described. This is the first observation of enzyme methemoglobinemia in Czech and Slovak populations. Key-words: methemoglobinemia, deficit cytochrome-b5-reductase
114 ÚVOD Methe oglo i é ie je o e o ě í harakterizova é příto ostí a or ál ě zvýše ého ožství ethe oglo i u MetH v krvi ad jeho f ziologi kou hra i í tj. ad %. Methe oglo i é ie ůže ýt vroze á e o získa á. O vroze é ethe oglo i é ii hovoří e zej é a v souvislosti s deficitem enzymu cytochrom-b5-reduktasy (Cb5R, EC , NADH-methemoglobinreduktasa, diaforasa, který se z % podílí a reduk i methemoglobinu. Defi it C R se dědí autoso ál ě re esiv ě a rozlišuje se dále a t p, podle toho zda postihuje pouze červe é krvi k t p I, e o vše h uňk t p II. V případě ethe oglo i é ie t pu I je hlav í he atologi ký projeve a óza MetH > %, která je v případě t pu II doprováze a e tál í retarda í a eurologi ký i symptomy. V ěkterý h případe h je tkáňová h po ie ko pe zová a ír ou pol té ií. Cb5R je flavoprotein s olekulovou h ot ostí kda. Jeho e rá ově váza á varia ta, jež se a hází v ito ho drií h e o e doplaz ati ké retikulu, stej ě tak jako solu il í er tro tár í varia ta, je kódová a ge e CYB5R3 (22q13- ter o sahují í e o ů; v případě solu il í for se epřepisuje část e o u a protei je tak o a i ok selin kratší. Je z á o přes růz ý h uta í v genu CYB5R3, z i hž ěkteré jsou společ é pro o a t p ethe oglo i é ie. Hereditár í ethe oglo i é ii spoje ou s deficitem C R je ut é odlišit předevší od po ěr ě vzá é hereditár í ethe oglo i é ie typu IV (deficit cytochromu b5, gen CYB5A a t pu M spojova é s výsk te he oglo i ů M, která je však autoso ál ě do i a t ího harakteru. V ěkterý h případe h je ethe oglo i é ie aso iová a s ji ý i vroze ý i poru ha i apř. příto ost nestabil ího he oglo i u, deficit glucosa-6-fosfátdehydrogenasy či p ruvátki as. Příči ou získa ý h ethe oglo i é ií polékový h je e pozi e e oge í h o ida tů a jeji h eta olitů a ti iotika, lokál í a estetika aj. MATERIÁL A METODIKA Sou or pa ie tů o sahoval 4 pacienty s ír ou resp. as pto ati kou ethe oglo i é ií (MetHb 7-11 %). V prvé případě se jed alo o leh e a oti kého letého hlap e s MetHb 7- % krev í o raz v or ě. Úči k MetH l reduková k s. askor ovou. Matka á MetHb v or ě, ote e í k dispozici. V druhé případě šlo o áhod ý zá h t leté že s MetH % ez kli i ký h příz aků. Třetí pa ie te lo ěsíč í děvče, které se
115 arodilo spo tá ě předčas ě v týd ů z I. gravidity (PH 2490 g, PD 46 cm), bez ikteru, ale cya oti ké se zvýše ou hladi ou MetH %. Pro opakova ý rozvoj d sp oe lo d a de hové podpoře. Po zaháje í terapie k s. askor ovou došlo u této pa ie tk k poklesu MetHb na 8- %. O a rodiče jsou as pto atičtí s MetHb v or ě. Čtvrtý pa ie te la rov ěž ěsíč í holčička aroze a spo tá ě v týd ů z IV. gravidit PH g, PD cm) s a ózou MetH, %. Na terapii k s. askor ovou došlo k postup é u poklesu hodnot MetH a %. O a rodiče i starší souroze i jsou as pto atičtí. Hemoglo i opatie la v louče a prostřed i tví testů tepel é sta ilit a elektroforéz he oglo i ů provede ý h podle sta dard í h postupů. Pro sta ove í aktivit C R la použita perifér í krev v hepari u, z íž la po odstra ě í plas e trifuga í připrave á u ěč á suspe ze v PBS o sahují í EDTA a erkaptoetha ol :. Aktivita C R la stanovena spektrofotometricky v te perova é s ěsi C po o í artifi iál ího su strátu ferrik a idu sledová í poklesu a sor a e při I fi ite Na oqua t, Tecan, Švý arsko. Spe ifi ká aktivita U/g H uvádě á v literatuře la vztaže a k ožství hemoglobinu v l zátu sta ove é rov ěž spektrofoto etri k při. Pro sekve ová í ge u CYB5R3 la použita ge o i ká DNA izolova á z perifér í krve EDTA) po o í QIAa p DNA kitu Qiage, USA. Vše h e o l a plifiková po o í PCR PCR pri er a pod í k jsou k dispozi i a v žádá í a sekve č í a alýza la provede a a sekve átoru ABI Pris Applied Bios ste, USA. VÝSLEDKY Ve vše h případe h la v louče a he oglo i opatie, defi it p ruvátki asy i glukosa-5- fosfátdeh droge asy. U prv ího pa ie ta lo prokázá o, že á s íže ou aktivitu C R, jež odpovídá % ko trol í h aktivit Ta.. Molekulár ě-ge eti ká a alýza potvrdila, že je heteroz gote pro z á ý pol orfiz us. G>A resp. p.pro Pro a ovou uta i (c.400a>g resp. p.met134val) v 5. exonu genu CYB5R3 (O r. A. As pto ati ká atka á or ál í aktivitu C R U/g H, ref. roz ezí:, -, U/g H a e í ositelkou ani jed é ze su stitu í. Druhá pa ie tka je heteroz gote pro su stitu i. G>A, tj. p.arg His v. e o u Ta., O r. A. Sta ove í aktivit C R e lo ož é zajistit. Třetí pa ie tka á a 2 % aktivitu Cb5R a je heterozygotem pro mutaci c.316g>a (p.val106met) ve 4. e o u Ta., O r. A. O a rodiče ají rov ěž s íže ou aktivitu C R - otec 3,4 U/g
116 Hb a matka 4,9 U/g Hb (8,42-, U/g H. Ote je ositele stej é uta e jako d era, u atk i přes s íže ou aktivitu C R e la uta e potvrzena. U čtvrté pa ie tk l deteková pokles aktivity Cb5R na 18 %. Sekve ová í lo potvrze o, že je dvojitý heterozygotem pro mutaci c.536c>a (p.ala179glu) a c.757g>a (p.val253met) v 6. a 9. exonu genu CYB5R3 (Tab. 1, Obr. 1A). O a rodiče ají s íže ou aktivitu C R otec 7,68 a matka 6,28 U/g Hb (8,42-13,66 U/g Hb), souroze i e li a al zová i. DISKUZE Muta e p.met Val detekova á u prv ího pa ie ta se a hází a C-konci beta-řetěz e FAD-vaze é do é O r. B, který je krátkou s čkou propoje s NAD(P)H-vazebnou do é ou. )á ě a ohla vést k osla e í ko taktu ezi o ě a do é a i a ovliv it tak sta ilitu e z u. Muta e p.met Val e la dosud popsá a. Su stitu e. G>A pro zá ě u p.arg His popsa ou u druhého pa ie ta je uvádě a v data ázi ( jako vzá ý pol orfis us; je situová a ve FAD-vaze é do é ě v lízkosti vaze ého ísta pro NAD(P)H a ohla epří o ovlivňovat i terak i ezi o ě a kofaktor. Muta e. G>A p.val Met potvrze á u třetího pa ie ta je stej ě jako v případě uta e p.met Val součástí FAD-vaze é do é a arušuje její h drofo í interakci (Obr. 1B). Vši h i pa ie ti jsou jed odu hý i heteroz got v kazují í i ír ou resp. asympto ati kou ethe oglo i é ii, takže v re esiv í ko figura i elze ez další h fu kč í h studií kauzalitu a t pové zařaze í detekova ý h uta í ge u CYB5R3 jed oz ač ě hod otit. Týká se to zej é a ově detekova é uta e (p.met134val) a vzá ého SNIPu. Muta e p.val Met la již popsá a v ho oz got í for ě u italského pa ie ta vedou í u ěho k ethe oglo i é ii t pu I. V případě uta e. C A p.ala Glu detekova é u. pa ie ta se jed á rov ěž o ovou uta i - la popsá a pouze uta e vedou í k zá ě ě Ala Thr afri kého, irského, ture kého a a eri kého původu [2,6,8] a Ala179Val thajského původu [9] způso ují í ethe oglo i é ii t pu I. Jed á se o uta i a házejí í se na C-konci NAD(P)H-vaze é do é y (Obr. 1B) ovlivňují í epří o vazbu NAD(P)H, a tí páde katal ti ké vlast osti e z u. V aše případě se tato uta e vyskytuje u pacienta v heteroz got í uspořádá í společ ě s uta í p.val Met. Jed á se o ejfrekve tova ější uta i v genu CYB5R3 leží í rov ěž v NAD(P)H-vaze é do é ě (Obr.
117 1B), která vede k aruše í ezido é ové i terak e a ted k z ě ě stéri ké ko for a e proteinu [6]. V ho oz got í for ě la tato uta e popsá a jak v souvislosti s ethe oglo i é ii t pu I polský, hola dský a ře ký původ [2,6,10], tak i typu II (u ě e kého pa ie ta [8]. I kd ž se předpokládá, že pa ie ti, kteří jsou jed odu hý i heteroz got, jsou o v kle as pto atičtí a s pto se o jeví až u ho oz gotů e o dvojitý h heteroz gotů, je toto rozděle í spíše o e ého harakteru. Větši a jedi ů s muta e i vedou í i k ethe oglo i é ii t pu I jsou as pto atičtí. Mez íke je spíše úroveň MetHb, kdy ho oz goti a dvojití heteroz goti s MetHb v roz ezí - 35 % se mohou projevovat a ózou popř. pol té ií. Další s pto se o v kle v sk tují až při árůstu MetH ad 35 % - duš ost, sla ost, dlo, eta oli ká a idóza, srdeč í ar t ie, kó a MetH 70 % je fatál í. U jed odu hý h heteroz gotů s MetH do %, je obvykle patr é pouze lehké z arve í kůže 11,12). )ÁVĚR Výsk t e z ové ethe oglo i é ie, která je e de i kého harakteru, je elosvětově po ěr ě vzá ý. S v sokou frekve í se v sk tuje převáž ě v a eri ké popula i i diá ů k e e Navajo a Čipejava ů ka adští do orod i) a u Jakutů a Si iřa ů (11,13,14). V Evropě l zaz a e á ojedi ělé případ irského 15, italského 16, ře kého, polského 10) i ě e kého původu ). Výše potvrze ý případ dvojitého heteroz gota s ethe oglo i é ií t pu I je prv í literár ě popsa ý případe e z ové ethe oglo i é ie v české a slove ské popula i. Na Slove sku l popsá pouze výskyt hemoglobinu M Saskatoon (17) souvisejí í s ethe oglo i é ií t pu M. Ni é ě jak je patr é z uvede ý h případů, lze v tě hto lokalitá h předpokládat výsk t zej é a as pto ati ký h heteroz got í h jedi ů itlivý h vůči e pozi i o idač í i látka i, které ohou vést k zhorše í projevů ethe oglo i é ie. PODÍL AUTORŮ NA RUKOPISU RM podíl a e z ový h a alýzá h, zpra ová í rukopisu,
118 MP a PK podíl a e z ový h a olekulár ě-ge eti ký h a alýzá h, připo í ková í rukopisu, DP, DS a KM léč a pa ie tů a připo í ková í rukopisu, VD připo í ková í rukopisu. PODĚKOVÁNÍ Prá e la podpoře a gra te IGA UP LF_2016_014. LITERATURA 1. Mansouri A, Lurie AA. Concise review: methemoglobinemia. Am J Hematol 1993; 42: Percy MJ, Lappin TR. Recessive congenital methaemoglobinaemia: cytochrome-b5- reductase deficiency. Brit J Haematol 2008; 141: Tomatsu S, Kobayashi Y, Fukumaki Y, Yubiscui T, Orii T, Sakaki Y. The organization and the complete nucleotide sequence of the human NADH-cytochrome b5 reductase gene. Gene 1989; 80: Bulíková A. A é ie. I : Neo kologi ká he atologie.. V dá í M. Pe ka, A. Bulíková et al.), Grada, Praha, Beutler E. Red cell metabolism: A manual of biochemical methods. 3rd edition. New York, USA, Grune & Straton, 1984; Dekker J, Eppink MHM, van Zwieten R, et al. Seven new mutation in the nicotinamide adenine dinucleotide reduce-cytochrome b5 reductase gene leading to methemoglobinemia type I. Blood 2001; 97: Shirabe K, Yubisui T, Borgese N, Tang Ch, Hultqiust DE, Takeshita M. Enzymatic instability of NADH-cytochrome b5 reductase as a cause of hereditary methemoglobinemia type I (red cell type). J Biol Chem 1992; 67: Kügler W, Pekrun A, Laspe P, Erdlenbruch B, Lakomek M. Molecular basis of recessive congenital methemoglobinemia, types I and II: exon skipping and three novel missense mutations in the NADH cytochrome b5 reductase (diaphorase 1) gene. Hum Mutat 2001; 17: 348.
119 9. Higaso K, Manabe J-I, Yubisui T, et al. Molecular basis of hereditary methaemoglobinaemia, types I and II: two novel mutations in the NADH-cytochrome b5 reductase gene. Br J Haematol 1998; 103: Grabowska D, Plochocka E, Jablonska-Skwiecinska A, et al. Compound heterozygosity of two missense mutations in the NADH cytochrome b5 reductase gene of a Polish patient with type I recessive congenital methaemoglobinaemia. Eur J Haematol 2003; 70: Prchal JT, Gregs XT. Red cell enzymes. Hematology 2005; 2005: Jaffe E R. Enzymopenic hereditary methemoglobinemia: a clinical/biochemical classification. Blood Cells 1986; 12: Balsamo P, Hardy WR, Scott EM. Hereditary methemoglobinemia due to diaphorase deficiency in Navajo Indians. J Pediatr 1964; 65: Scott EM, Lewis M, Kaita H, Chown B, Giblett ER. The absence of close linkage of methemoglobinemia and blood group loci. Am J Hum Genet 1963;15: Percy MJ, Crowley LJ, Davis CA, et al. Recessive congenital methaemoglobinaemia: functional characterization of the novel D239G mutation in the NADH-binding lobe of cytochrome b5 reductase. Br J Haematol : Fermo E, Bianchi P, Vercellati C, et al. Recessive hereditary methemoglobinemia: two novel mutations in the NADH-cytochrome b5 reductase gene. Blood Cells Mol Dis 2008; 4: Divoký V, Wal z skova S, Pospíšilová D, et al. Některé vzá ější for hereditár í h a é ií v sk tují í se v dospělé popula i v ČR a SR β talase ie a esta il í he oglo i ové varia t. V itř lék ; : ADRESA Mgr. Re áta Mojzíková, Ph.D. Ústav iologie LF UP H ěvotí ská, Olomouc r.mojzikova@gmail.com
120 Ta.. Pa ie ti se s íže ou aktivitou C R io he i ká a olekulár ě-ge eti ká charakterizace. Pacient 1 Pacient 2 Pacient 3 Pacient 4 Věk/pohlaví 15/M /Ž /Ž /Ž MetHb (%) Cb5R (U/g Hb) 3,0 n.d. 2,44 2,04 Mutace 400A>G 890G>A 316G>A 536C>A/757G>A Exon ,9 AK zá ě a Met134Val Arg297His Val106Met Ala179Glu/Val253Met Poz.: : věk v ěsí í h; M/Ž: už/že a;.d.: esta ovová o; ref. roz ezí pro aktivitu Cb5R: 8,42-13,66 U/g Hb); pacient 1,3 a 4 na terapii kys. askorobovou; oz ače í uta e dle HGMD gen CYB5R3 (22g13.2, NM_ ).
121 LEGENDY K OBRÁ)KŮM Obr. 1. A) Mutace v genu CYB5R3 pacient 1: c.132g>a (p.pro44pro, exon 2) a c.400g>a (p.met134val, exon 5); pacient 2: c.890g>a (p.arg297his, exon 9); pacient 3: c.316g>a (p.val106met, exon 4) a pacient 4: c.536c A (p.ala179glu, exon 6) a c.757g A (p.val253met, exon 9); B A i ok seli ová sekve e lidské NADH-Cb5R (ref. sekvence NM_000398, 301 aminokyselin) (2) s lokaliza í detekova ého pol orfis u a a i ok seli ový h zá ě žlutě. FAD a NAD P H-vaze á do é a jsou v z ače é odře a červe ě 6.
122
123 Příloha 3 : Talasemie (zahraniční publikace s IF) Divoka, M., Partschova, M., Kucerova, J., Mojzikova, R., Cermak, J., Pospisilova, D., Fabryova, V., Prochazkova, D., Indrak, K.,Divoky, V. Molekular Characterization of β- Thalassemia in the Czech and Slovak populations: Mediterranean, Asian and Unique Mutations. Hemoglobin. Hemoglobin Jun; 40(3): IF 0.787
 Journal homepage: http://www.tandfonline.")


: Molecular Characterization of β-thalassemia in the Czech and Slovak Populations:")



124 Hemoglobin international journal for hemoglobin research ISSN: (Print) X (Online) Journal homepage: Molecular Characterization of β-thalassemia in the Czech and Slovak Populations: Mediterranean, Asian and Unique Mutations Martina Divoka, Martina Partschova, Jana Kucerova, Renata Mojzikova, Jaroslav Cermak, Dagmar Pospisilova, Viera Fabryova, Daniela Prochazkova, Karel Indrak & Vladimir Divoky To cite this article: Martina Divoka, Martina Partschova, Jana Kucerova, Renata Mojzikova, Jaroslav Cermak, Dagmar Pospisilova, Viera Fabryova, Daniela Prochazkova, Karel Indrak & Vladimir Divoky (2016): Molecular Characterization of β-thalassemia in the Czech and Slovak Populations: Mediterranean, Asian and Unique Mutations, Hemoglobin, DOI: / To link to this article: View supplementary material Published online: 09 Mar Submit your article to this journal View related articles View Crossmark data Full Terms & Conditions of access and use can be found at Download by: [Knihovna Univerzity Palackeho] Date: 10 March 2016, At: 06:40
125 ISSN: (print), X (electronic) Hemoglobin, Early Online: 1 7! 2016 Taylor & Francis. DOI: / ORIGINAL ARTICLE Molecular Characterization of b-thalassemia in the Czech and Slovak Populations: Mediterranean, Asian and Unique Mutations Downloaded by [Knihovna Univerzity Palackeho], [${individualuser.displayname}] at 06:40 10 March 2016 Martina Divoka 1, Martina Partschova 2, Jana Kucerova 2, Renata Mojzikova 2, Jaroslav Cermak 4, Dagmar Pospisilova 3, Viera Fabryova 5, Daniela Prochazkova 6, Karel Indrak 1, and Vladimir Divoky 1,2 1 Department of Hemato-Oncology, Faculty of Medicine and Dentistry, Palacky University and University Hospital, Olomouc, Czech Republic 2 Department of Biology, Faculty of Medicine and Dentistry, Palacky University, Olomouc, Czech Republic 3 Department of Pediatrics, Faculty of Medicine and Dentistry, Palacky University and University Hospital, Olomouc, Czech Republic 4 Institute of Hematology and Blood Transfusion, Prague, Czech Republic 5 Department of Hematology and Biochemistry, St. Michael Hospital, Bratislava, Slovak Republic and 6 Department of Pediatrics, Masaryk Hospital, Usti nad Labem, Czech Republic Abstract b-thalassemia (b-thal) is considered rare in Central Europe. As in other malaria-free regions, the presence of b-thal in Central Europe reflects historical and recent immigration, and demographic changes that have influenced the genetic variability of the current populations living in this area. This study assesses the frequency and spectrum of mutations on the b-globin gene in Czech and Slovak subjects with clinical symptoms of thalassemia. The results of the initial part of this research were published more than two decades ago; the aim of this study was to update these original reports. During the period from 2002 to 2015, 400 cases from Czech and Slovak hematological centers were analyzed. Twenty-nine b-thal mutations, identified in 356 heterozygotes from 218 unrelated families, involve five unique mutations including a recently described insertion of a transposable L1 element into the b-globin gene. One mutation described here is reported for the first time. Most of the mutations were of Mediterranean origin and accounted for 82.0% of cases. All but one case studied were heterozygous carriers, manifesting b-thal minor, with rare exceptions represented by the rare (b 0 ) codons 46/47 (+G) (HBB: c.142_142dupg) mutation associated with an a-globin gene quadruplication and by dominantly inherited b-thal with a more severe phenotype. One double heterozygous b-thal patient was a recent immigrant from Moldavia. The list of db-thal alleles (26 carriers, 16 families) contains Hb Lepore and two types of db 0 -thal deletions. In the past, genetic drift and migration as well as recent immigrations were responsible for the introduction of Mediterranean alleles, while several mutations described in single families were of local origin. Introduction Hemoglobinopathies are known to occur at low frequencies in the populations of West, North and Central European countries (1). Reports describing thalassemia or hemoglobin (Hb) variants from malaria-free countries often bring novel insights into the epidemiology of these genetic disorders (2 10) and contribute to the understanding of the pathophysiology of these diseases (11,12) or might even represent novel mechanisms of these congenital defects (13 15). In this report, we have updated the molecular analysis of b- and db-thalassemia (b- and db-thal) alleles found in the Czech and Slovak populations. Since the original report (4), Address correspondence to Dr. Vladimir Divoky, Department of Biology, Faculty of Medicine and Dentistry, Palacky University, Hnevotinska 3, Olomouc, CZ , Czech Republic. Tel: Fax: vladimir.divoky@upol.cz Keywords b-thalassemia (b-thal), genetic epidemiology, isolated unique mutations, Mediterranean alleles History Received 22 November 2015 Revised 17 December 2015 Accepted 20 December 2015 Published online 4 March 2016 and several other previous reports describing unique mutations (16 18), the number of b-thal chromosomes has more than tripled and the spectrum of b-thal mutations has risen considerably. The relatively high frequency of b-thal alleles, mostly specific to the Mediterranean Basin, reported up to now in the Czech and Slovak populations (4,19,20) has been attributed to a long common history between Czechs and Slovaks and some Mediterranean populations in the multinational Habsburg monarchy, and later in the Austrian- Hungarian monarchy. Other historical influences such as Ottoman Turk invasions of these lands during the 16th and 17th centuries (21,22) are also a factor. Here, we have summarized the results obtained from 2002 to 2015 for families with b- and db-thal chromosomes living in the Czech and Slovak Republics (former Czechoslovakia); the subjects were mostly of native Czech or Slovak origin. Besides the four previously described unique mutations
126 2 M. Divoka et al. Hemoglobin, Early Online: 1 7 Downloaded by [Knihovna Univerzity Palackeho], [${individualuser.displayname}] at 06:40 10 March 2016 originating in local populations (15 18), one novel mutation was identified and was added to the existing HbVar database ( (23). Materials and methods In a series of 400 cases from 240 families, hereditary hypochromic microcytic anemia and elevated Hb A 2 and/or Hb F values suggested a b- or db-thal determinant. The patients samples were collected and referred to us for genetic analysis between 2002 and The Ethics Committee of Palacky University Hospital, Olomouc, Czech Republic approved the study and informed consent was obtained from all individuals. Blood sample measurements (using Sysmex XE 5000 analyzer; Sysmex, Kobe, Japan), Hb electrophoresis, Hb A 2 and Hb F quantification have been described in previous reports (24,25). Genomic DNA was isolated from peripheral blood samples collected into vacutainers containing EDTA, using either phenol/chloroform/isoamyl alcohol extraction with ethanol precipitation, the Puregene Õ Blood Core Kit B (Qiagen Inc., Valencia, CA, USA) or an automatic DNA analyzer MagNA Pure (Roche Diagnostics GmbH Deutschland, Mannheim, Germany). The polymerase chain reaction (PCR) primers and conditions used to amplify the b-globin gene including the 5 0 and 3 0 untranslated regions (5 0 and 3 0 UTRs) (NM_ ) are available upon request. The PCR products were purified with a QIAquick PCR purification kit (Qiagen Inc.) and used for dot-blot analysis of amplified DNA using 32 P-labeled oligonucleotide probes (4) or for direct PCR product sequencing in both directions with a BigDye terminator kit (Applied Biosystems, Foster City, CA, USA) using an ABI PRISMÔ 310 or 3100 Genetic Analyzer (Applied Biosystems). In addition, several db-thal alleles were characterized through MLPA (multiplex ligationdependent probe amplification) (SALSA MLPA probemix P102-B2 HBB; MRC-Holland, Amsterdam, The Netherlands). Results During the period from 2002 to 2015, 400 subjects with features of b-ordb-thal traits were analyzed; the b-ordb-thal alleles were detected in 382 carriers from 234 unrelated families. One patient was a double heterozygote for b-thal mutations. The cumulative results of the incidence of individual mutations together with their ethnic distribution are provided in Table 1. The Mediterranean mutation IVS-I-1 (G4A) (HBB: c G4A) was present in about onefourth of Czech and Slovak families and thus remains the most frequent b-thal mutation in this population, as previously described (4). In earlier studies performed in Czech and Slovak families, the mutation was associated with haplotype Va [in six families, in which haplotype analysis was carried out (19,26)] suggesting this mutant allele in the affected Czech and Slovak families originated in the Mediterranean. Other common Mediterranean mutations detected previously on Mediterranean haplotypes in Czech and Slovak families (19) (Supplementary Table 1) and found in more than 10 Czech or Slovak families in this study were: IVS-I-110 (G4A) (HBB: c.93 21G4A); IVS-I-6 (T4C) (HBB: c T4C), codon 39 (C4T) (HBB: c.118c4t) and IVS-II-1 (G4A) (HBB: c G4A). In total, 293 subjects from 184 families (82.0% of subjects, 84.4% of families) carry b-thal alleles previously reported from the endemic Mediterranean region (Figure 1). In addition, all db-thal alleles detected are common in Mediterranean countries. The second most common subgroup of mutations (in terms of their origin) are those which originated locally (Figure 1). Of these, the codons 38/39 ( C) (HBB: c.118delc) mutation (18) is relatively frequently found only in the Czech population. Another unique mutation, codon 112 (T4A) (HBB: c.339t4a), was observed in one Slovak family in our cohort and was likely related to the original case described earlier (16). The insertion of a transposable element causing b + -thal in a Ukrainian mother and daughter living in Slovakia has been described separately (15). Table 2 shows a comparison of hematological data for two unique and one rare b 0 -thal mutations detected in single families, the codons 7/8 (+G) (HBB: c.24_24dupg) mutation has already been described in a Czech language journal (17), and codons 46/47 (+G) (HBB: c.142_142dupg) in one Italian patient from Sicily (27), while the codon 52 ( A) (HBB: c.158dela) mutation is reported here for the first time. The hematological values for the three family members with the codons 46/47 mutation were more severe than expected for b 0 -thal heterozygotes and a search for another determinant (responsible for this severe phenotype) revealed an a-globin gene quadruplication in all three patients (Supplementary Figure 1). However, except for more pronounced microcytic anemia and splenomegaly detected in one child (Table 2), the other affected family members did not exhibit severe clinical symptoms (signs of hemolytic anemia with splenomegaly) and none of the patients required transfusions. The molecular defects of 18 cases with suspected b- or db-thal traits (i.e. familiar microcytosis, hypochromia, borderline to elevated Hb A 2 and/or Hb F, normal serum ferritin level) remain unknown, despite extensive sequence analysis involving the flanking regions of the b-globin gene as well as MLPA analysis of the b-globin gene cluster. Discussion b-thalassemia is considered rare in Central Europe (1). As in other non malaria regions, the presence of b-thal in Central Europe reflects the historical immigration and demographic changes that have influenced the genetic variability of the current populations living in this area (28). The Czech Republic (Bohemia and Moravia) was originally populated by Celts and invaded by Germanic tribes. The Slavs become the dominant population from the 6th to 8th centuries, surrounded by Germans (22). The Slovaks were Slavs who settled to the east of Moravia. Early on in their history, they fell under the rule of the Magyars, or Hungarians, who were Finno- Ugric in ethnic origin rather than Slavic (21). Even though close contact between the Czechs and Slovaks came much later (from 1620 but mainly after 1918), the resulting gene-flow is likely to be responsible for a very similar thalassemia allele frequency in the Czechs and Slovaks. Despite some cultural differences, the Czechs and Slovaks shared political and historical regions that emerged in Central Europe long enough
127 DOI: / Thalassemia in the Czech and Slovak Populations 3 Table 1. The incidence of b-thalassemia and db-thalassemia mutations in Czech and Slovak populations found during the period. Downloaded by [Knihovna Univerzity Palackeho], [${individualuser.displayname}] at 06:40 10 March 2016 b-thalassemia Mutations HbVar ID HGVS Nomenclature a (HBB:) Families/ Cases Relative Frequency (% of chromosomes) Type of Thalassemia b ; Ethnic Background b (possible haplotype) c IVS-I-1 (G4A) 817 c G4A 53/ b 0 ; Mediterranean (Va) IVS-I-110 (G4A) 827 c.93 21G4A 40/ b + ; Mediterranean (I; IX) IVS-I-6 (T4C) 826 c T4C 27/ b ++ ; Mediterranean (VI; X) codon 39 (C4T) 845 c.118c4t; p.gln40* 23/37 d 10.3 b 0 ; Mediterranean (I) IVS-II-1 (G4A) 884 c G4A 15/ b 0 ; Mediterranean codons 38/39 ( C) 843 c.118delc; p.gln40argfs*22 7/ b 0 ; Czech (II) codon 8 ( AA) 785 c.25_26delaa; p.lys9valfs*14 7/12 d 3.3 b 0 ; Mediterranean IVS-II-745 (C4G) 891 c C4G 7/ b + ; Mediterranean (VIIa) 87 (C4T) 759 c. 137C4T 6/7 2.0 b ++ ; German/Italian codon 121 (G4T) 951 c.364g4t; p.glu122* 4/12 e 3.3 b 0 ; Caucasian/ Czech-Slovak? (Va) codons 41/42 ( TTCT) 849 c.126_129delcttt; 4/4 1.1 b 0 ; Asian (Chinese) p.phe42leufs*19 codons 82/83 ( G) 875 c.251delg; p.gly84alafs*6 3/8 2.2 b 0 ; Azerbaijani polya (AATAAA4AATGAA) 969 c.*111a4g 3/4 1.1 b ++ ; Mediterranean 87 (C4G) 758 c. 137C4G 2/3 0.8 b ++ ; Mediterranean codon 17 (A4T) 800 c.52a4t; p.lys18* 2/2 0.6 b 0 ; Asian (Chinese) 86 (C4A) 760 c. 136C4A 2/2 0.6 b ++ ; Italian codons 46/47 (+G) 2974 c.142_142dupg; 1/3 0.8 b 0 ; Italian (see also Table 2) p.asp48glyfs*6 IVS-I-5 (G4C) 824 c G4C 1/3 0.8 b + ; Mediterranean codon 52 ( A) f 2973 c.158dela; p.asp53valfs*9 1/3 0.8 b 0 ; Czech (see also Table 2) codon 112 (T4A) 948 c.339t4a; p.cys113* 1/2 e 0.6 b 0 ; Slovak codons 7/8 (+G) f,g 2972 c.24_24dupg; p.lys9glufs*15 1/2 0.6 b 0 ; Slovak (IX; see also Table 2) 44 bp deletion 975 c.76_ del 1/2 0.6 b 0 ; Greek codon 5 ( CT) 783 c.17_18delct; p.pro6argfs*17 1/2 0.6 b 0 ; Mediterranean IVS-II-765 L1 h 2892 L1 insertion between 1/2 0.6 b + ; Ukrainian c _ codon 45 ( T) 855 c.138delt; p.phe46leufs*16 1/2 0.6 b 0 ; Asian (Pakistani) codon 37 (TGG4TGA) 841 c.114g4a;p.trp38* 1/1 0.3 b 0 ; Asian (Saudi Arabian) 619 bp deletion 979 c _*342delinsAAGTAGA i 1/1 0.3 b 0 ; Indian codon 6 ( A) 784 c.20dela; p.glu7glyfs*13 1/1 0.3 b 0 ; Mediterranean codons 71/72 (+A) 869 c.216_217insa; p.ser73lysfs*2 1/1 0.3 b 0 ; Asian (Chinese) Total 218/ db-thalassemia Mutations Hb Lepore j HBD:HBB: fusion 11/ db fusion Hb Sicilian 1035 NG_ :g. 4/ (db) 0 -thalassemia 64336_77738del Macedonian/Turkish 1036 HBB:HBD: complex 1/1 3.8 (db) 0 -thalassemia rearrangement Total 16/ a HBB accession NM_ b From HbVar [ (23)], IthaGenes [( (32)] and from (54). c Based on haplotype studies performed on a limited number of families published in a Czech language journal (19). Haplotype analysis was performed using a PCR-RFLP method (56) at nine polymorphic sites: HincII-5 0 e, XmnI-5 0 g, HindIII- G g, HindIII- A g, HincII- b, HincII-3 0 b, HinfI-5 0 b, AvaIIb and BamHI-3 0 b, and the haplotypes were numbered according to (35). For more information, see the Supplementary Table 1. d One patient of Moldavian origin was a compound heterozygote for codon 8/codon 39 with b-thal major. e Likely relatives of previously described families/cases (4,16). f Newly described mutation. g First described in a Czech language journal (17). h Described separately (15). i Sequence verified as described (57) not as NG_ :g.71609_72227del619, the current HbVar annotation of 619 bp deletion HBB variant. j Precisely specified cases were either Hb Lepore-Baltimore (d 50 /b 86 ; NG_ :g.63564_70978del; HbVar ID: 743) (n ¼ 3) or Hb Lepore-Boston- Washington (Hb LBW, d 87 /b 116 ; NG_ :g.63632_71046del; HbVar ID: 744) (n ¼ 3). that many families with Czech ancestry contain members of Slovak origin and vice versa, which makes it difficult to distinguish unique and common mutations between the Czech and Slovak populations. For this purpose we consider the Czech and Slovak Republics as one region. Formerly, the frequency of b-thal mutations in Czechoslovakia (now the Czech and Slovak Republics) was described as specifically different from that found in the Mediterranean and Balkan countries; the main difference was attributed to a seemingly high relative frequency (45.2%) of the IVS-I-1 allele among the Czech and Slovak thalassemia carriers (4), and these data were also cited in the following comparative studies (29). The IVS-I- 1 mutation was even proposed to have originated in Eastern Europe (30). However, these data were based on a relatively small number of b-thal chromosomes analyzed from the former Czechoslovakia. Here, in a much larger study allowing more reliable comparison, the relative frequency of the most common b-thal mutations does not seem to differ from certain
![4 M. Divoka et al. Hemoglobin, Early Online: 1 7 Downloaded by [Knihovna Univerzity Palackeho], [${individualuser.displayname}] at 06:40 10 March 2016 Figure 1.](/docs-images/105/176107544/images/128-0.jpg "Summary of frequencies of b-thal chromosomes in the Czech and Slovak populations according to their ethnic origin.")
128 4 M. Divoka et al. Hemoglobin, Early Online: 1 7 Downloaded by [Knihovna Univerzity Palackeho], [${individualuser.displayname}] at 06:40 10 March 2016 Figure 1. Summary of frequencies of b-thal chromosomes in the Czech and Slovak populations according to their ethnic origin. The relative frequencies of b-thal chromosomes in individual ethnic categories were calculated from the b-thal chromosome frequencies as depicted in Table 1. The ethnic origin/distribution data for individual b-thal mutations were compiled from HbVar [ globin.cse.psu.edu/hbvar (23)] and IthaGenes [ (32)] databases and one review article (54). The Asian category of mutations involves rarely represented Chinese, Pakistani, Saudi Arabian and Indian mutations (see Table 1). Table 2. Hematological data of one rare and two unique b-thalassemia mutations. Mutation Patients Sex-Age Hb (g/dl) RBC (10 12 /L) MCV (fl) MCH (pg) Hb A 2 (%) Hb F (%) Mediterranean populations. The incidence of the IVS-I-1 allele (27.1% of molecularly characterized b-thal cases in this study) is similar to that observed in some Mediterranean regions, such as the Iberian Peninsula (31), some parts of Italy (27) or some population groups (Lebanese Christians) (30); a comparison of IVS-I-1 allele s relative frequencies for different populations/ regions is available at (32). Although found in association with different haplotypes (33,34), the vast majority of the IVS-I-1 allele observed in Mediterranean regions was linked with haplotype V [ psu.edu/hbvar; the original haplotype designation associated with IVS-I-1 was described as the Mediterranean subhaplotype Va (35)]. As the same subhaplotype was previously identified for the Czech IVS-I-1 alleles (19,26), it is very likely that this mutation was introduced to the Czech and Slovak populations from the Mediterranean Basin as we proposed (4,20) and did not originate in Eastern Europe as ascribed (30). The second most common mutation in our cohort, IVS-I-110 (G4A) (17% of molecularly characterized b-thal cases in this study), is the most common b-globin gene mutation in Greece (36), and is also common in several other Mediterranean populations [documented in the HbVar Reticulocytes (%) codons 7/8 (+G) a mother F NA son M NA codon 52 ( A) four F-9, F NA relatives b M-34, M-29 codons 46/47 (+G) c mother F son M daughter d F Hb: hemoglobin; RBC: red blood cell count; MCV: mean corpuscular volume; MCH: mean corpuscular Hb; normal control range for reticulocytes: %; NA: not analyzed. a From (17). b Range of values. c All family members with the codon 46/47 mutation also carry the a-globin gene quadruplication (see Supplementary Figure 1). d The only family member presenting with splenomegaly. database ( or in the IthaGenes database ( The haplotypes previously associated with this mutation in selected Czech/ Slovak families were I and IX (19) (Supplementary Table 1) the most common haplotypes associated with this mutation in the Mediterranean (35). Also, the spectrum of other b-thal determinants of Mediterranean origin reported in this study further supports our original conclusion that the majority of b-thal genes in the Czech and Slovak populations were imported from the Mediterranean (4). Similar results were published for the indigenous Hungarian and German populations (5,37). It seems likely that the occurrence of these mutations corresponds with a common history (400 years) for Czechs, Slovaks, Germans and Hungarians as well as some Mediterranean populations joined from 1526 to 1918 to the Habsburg monarchy and later to the Austrian-Hungarian monarchy. In some subjects, namely one family with codon 39, one with codon 8 ( AA), another with the Greek 44 bp deletion and one family carrying Hb Lepore, ancestors from the Mediterranean region were identified, suggesting the ancestors of the individuals affected had recently migrated to
129 DOI: / Thalassemia in the Czech and Slovak Populations 5 Downloaded by [Knihovna Univerzity Palackeho], [${individualuser.displayname}] at 06:40 10 March 2016 the Czech Republic (e.g. a group of Greeks settled in the former Czechoslovakia after the Second World War). However, the vast majority of b-thal-affected families did not report Mediterranean relatives in the two preceding generations, indicating that the mutant genes were introduced into the Czech and Slovak populations during the Austrian- Hungarian empire or earlier. In industrialized Northern and Western European countries, b-thal and other hemoglobinopathies have recently become much more common through high immigration rates from endemic areas (10,28,38). Although immigration to the Czech and Slovak Republics is rising, it is not yet that high, and the presence of some mutations (such as the Turkish codon 8 mutation) in several families without a clear Mediterranean or Turkish background may be remnants of historical rather than recent events the regions were invaded by Huns (4th century) and Avars (6th to 8th centuries) and overrun by other Asian tribes such as the Mongols or Tartars (21,22). Two Asian mutations, codons 41/ 42 ( TTCT) and codon 17 (A4T), detected in six families without any obvious Asian influence on their pedigrees, may also have a historical Asian origin; both these mutations are common in the Far East (1). From the five mutations so far found only in local (non immigrant) families, the distribution of the codons 38/39 mutation is quite interesting. Most of the affected families come from the same region, central Moravia (the eastern part of the Czech Republic). Previous haplotype analysis of three of these families showed that the mutated allele was linked to haplotype II (19). These data suggest that this mutation may have originated relatively recently and due to a single founding event it appears to be regionally specific. The other unique b-thal mutation, the insertion of a transposable L1 element into the b-globin gene (depicted in Table 1 as IVS-II-765 L1, HbVar ID: 2892), represents a novel etiology of b-thal due to the silencing effect of repressive chromatin associated with a retrotransposon insertion (15). Similar to what was published for other non malaria countries [reviewed in (39)], b-thal mutation diversity in the Czech and Slovak populations has been increased by isolated unique mutations. One such mutation, codon 52, is described here for the first time and an additional one was described only in a Czech language journal (codons 7/8); both have now been added to the online HbVar database ( globin.cse.psu.edu/hbvar). In the case of the rare codons 46/ 47 mutation, the heterozygotes described in this study coinherited two extra a-globin genes (quadruplication of the HBA2 gene) that resulted in a more severe b-thal phenotype (more severe than would be expected for b 0 -thal heterozygotes), as described for other b 0 -thal carriers with such an a-globin genotype (40,41). Increased b-thal severity by a-globin gene triplication has already been published for a Czech patient (26), verifying the presence of these a-globin genotypes with an increased number of functional a-globin genes in the Czech population. Of some special interest are two other rare mutations, namely codons 82/83 ( G) (HBB: c.251delg) and codon 121 (G4T) (HBB: c.364g4t). The rare frameshift mutation at codons 82/83, originally described in Azerbaijanis (42,43), has been reported with a relatively high frequency from Germany (7). The origin of this mutation in Czech, Slovak [this report and (4)] and German families will remain unresolved until the haplotype analyses become available. It is possible, however, that the mutation was imported to Central Europe by Azerbaijanis, as it was suggested that this mutation might be specific for the population of Azerbaijan (44). The codon 121 mutation was originally described as a cause of severe hemolytic anemia with splenomegaly, called dominantly inherited inclusion body b-thal (11,45,46). However, later the mutation was associated with a variable clinical manifestation in heterozygotes among the families affected and even among carriers from the same family (47,48); the same was true for individuals from the Czech and Slovak families; the clinical picture of most carriers resemble b-thal traits associated with recessive mutants [this report and (4)]. All the patients we studied had four a-globin genes and sequencing analysis of both a1 and a2 genes from two selected individuals with an asymptomatic clinical picture failed to detect any abnormality (not shown). Thus, other recently identified modifiers of the clinical severity of b-thal (12,40,49,50) might be responsible for this variable clinical manifestation. Previous haplotype analysis in one Slovak and two Czech families with a codon 121 mutation revealed restriction fragment length polymorphism (RFLP) characteristics of haplotype Va (19), however, differences in the polymorphisms were recently observed through sequencing analysis of the (AT) X (T) Y repeat motif 5 0 to the b-globin gene (unpublished data, not shown). Interestingly, two of four Dutch families with the codon 121 mutation revealed the same haplotype (or at least the same 5 0 subhaplotype, referred to as h2) (48). Although we cannot exclude the possibility that (at least some of) the mutations arose from the same event and that the differences of the (AT) X (T) Y polymorphisms may have resulted from recombination at the recombination hot-spot, the rarity of this mutation rather supports the hypothesis that the causative codon 121 mutations are of de novo origin in individual European populations, as suggested for this type of mutation in non malaria countries with an otherwise low thalassemia frequency (39). Based on the above mentioned clinical and genetic analyses, we conclude that the only true dominantly inherited b-thal present in the Czech population is represented by the hyperunstable Hb variant described as Hb Hradec Kralove (Hb HK, HBB: c.347c4a) (51). An independent study involving a cohort of 70 b-thal carriers from the Slovak Republic (20) revealed mutations also represented in a cohort of this study. However, there are a few mutations not detected by us but described by others for Czech or Slovak b-thal subjects, which further increase the diversity of b-thal mutations in the Czech/ Slovak ethnicities; these were the Czech 4.2 kb b-thal deletion [HbVar ID: 983 (52)] and the polyadenylation (polya) signal AATAAA4GATAAA mutation [IthaID: 271 (53)]. Eighteen cases in our cohort with clinical features of b-thal trait but with no detectable mutation in the b-globin gene suggest that mutations in trans-acting factors causing the b-thal phenotype (54) remain to be determined in the indigenous Czech and Slovak populations. In fact, one Krüppel-like factor 1 (KLF1) gene mutation associated with increased Hb F levels has recently been identified in a Slovak individual (55).
130 6 M. Divoka et al. Hemoglobin, Early Online: 1 7 Downloaded by [Knihovna Univerzity Palackeho], [${individualuser.displayname}] at 06:40 10 March 2016 Acknowledgments The authors are indebted to all physicians from the Czech Republic and the Slovak Republic who participated in this study. Declaration of interest This study was supported by grant NT13587 (from the Ministry of Health Czech Republic), by Education for Competitiveness Operational Programme of the MSMT CR project CZ.1.07/2.3.00/ and by student projects IGA_LF_2015_015 and IGA_LF_2015_001 (from the Palacky University, Olomouc, Czech Republic). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article. References 1. Loukopoulos D, Kollia P. Worldwide distribution of b thalassemia. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, Eds. Disorders of Hemoglobin Genetics, Pathophysiology, and Clinical Management. New York, NY: Cambridge University Press. 2001: Laig M, Pape M, Hundrieser J, Flatz G. Mediterranean types of b-thalassemia in the German population. Hum Genet. 1990; 85(1): Hall GW, Barnetson RA, Thein SL. b Thalassaemia in the indigenous British population. Br J Haematol. 1992;82(3): Indrak K, Brabec V, Indrakova J, et al. Molecular characterization of b-thalassemia in Czechoslovakia. Hum Genet. 1992; 88(4): Ringelhann B, Szelenyi JG, Horanyi M, et al. Molecular characterization of b-thalassemia in Hungary. Hum Genet. 1993; 92(4): Heusterspreute M, Derclaye I, Gala JL, et al. b-thalassaemia in indigenous Belgian families: Identification of a novel mutation. Hum Genet. 1996;98(1): Vetter B, Schwarz C, Kohne E, Kulozik AE. b-thalassaemia in the immigrant and non-immigrant German populations. Br J Haematol. 1997;97(2): Maciag M, Plochocka D, Adamowicz-Salach A, et al. Diversity of thalassemia variants in Poland screening by real-time PCR. Acta Haematol. 2008;120(3): Van Zwieten R, Veldthuis M, Delzenne B, et al. Hemoglobin analyses in The Netherlands reveal more than 80 different variants including six novel ones. Hemoglobin. 2014;38(1): Kohne E, Kleihauer E. Hemoglobinopathies: A longitudinal study over four decades. Dtsch Arztebl Int. 2010;107(5): Thein SL. Dominant b thalassaemia: Molecular basis and pathophysiology. Br J Haematol. 1992;80(3): Mojzikova R, Dolezel P, Pavlicek J, et al. Partial glutathione reductase deficiency as a cause of diverse clinical manifestations in a family with unstable hemoglobin (Hemoglobin Hana, b63(e7) His-Asn). Blood Cells Mol Dis. 2010;45(3): Barbour VM, Tufarelli C, Sharpe JA, et al. a-thalassemia resulting from a negative chromosomal position effect. Blood. 2000; 96(3): Tufarelli C, Stanley JA, Garrick D, et al. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet. 2003;34(2): Lanikova L, Kucerova J, Indrak K, et al. b-thalassemia due to intronic LINE-1 insertion in the b-globin gene (HBB): Molecular mechanisms underlying reduced transcript levels of the b-globin(l1) allele. Hum Mutat. 2013;34(10): Divoky V, Gu L-H, Indrak K, et al. A new b 0 -thalassaemia nonsense mutation (codon 112, T!A) not associated with a dominant type of thalassaemia in the heterozygote. Br J Haematol. 1993;83(3): Kynclova E, Divoky V, Kovarikova L, et al. [New b 0 -thalassaemic insertion mutation (CD 7/8, +G) in a Slovak family, associated with the Mediterranean haplotype IX]. Vnitr Lek. 1999;45(3): Indrak K, Indrakova J, Kutlar F, et al. Compound heterozygosity for a b 0 -thalassemia (frameshift codons 38/39; C) and a nondeletional Swiss type of HPFH (A!C at NT 110, G g) in a Czechoslovakian family. Ann Hematol. 1991;63(2): Kynclova E, Kovarikova L, Fajkosova P, et al. [Haplotypes of the b-globulin locus in Czechs and Slovaks with b-thalassemia and structurally variant hemoglobins]. Vnitr Lek. 1998; 44(9): Fabryova V, Babusik P, Laluhova-Striezencova Z, et al. Nineteen years study of b-thalassaemia in Slovakia. Cent Eur J Public Health. 2012;20(4): Alexander J. Slovakia. In: Frucht RC, Ed. Eastern Europe: An Introduction to the People, Lands, and Culture, Vol. 2. Santa Barbara, CA: ABC-CLIO. 2005: Miller D. The Czech Republic. In: Frucht RC, Ed. Eastern Europe: An Introduction to the People, Lands, and Culture, Vol. 2. Santa Barbara, CA: ABC-CLIO. 2005: Giardine B, Borg J, Viennas E, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42(Database issue):d1063 D1069. ( globin.cse.psu.edu). 24. Kutlar A, Huisman THJ. Detection of hemoglobinopathies. In: Hommes FA, Ed. Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual. New York, NY: Wiley-Liss. 1991: Urrechaga E, Borque L, Escanero JF. The role of automated measurement of RBC subpopulations in differential diagnosis of microcytic anemia and b-thalassemia screening. Am J Clin Pathol. 2011;135(3): Indrak K, Fei Y-J, Li H-W, et al. A Czechoslovakian teenager with Hb E-b 0 -thalassemia [IVS-I-1 (G!A)] complicated by the presence of an a-globin gene triplication. Ann Hematol. 1991; 63(1): Giambona A, Vinciguerra M, Cannata M, et al. The genetic heterogeneity of b-globin gene defects in Sicily reflects the historic population migrations of the island. Blood Cells Mol Dis. 2011; 46(4): Modell B, Darlison M, Birgens H, et al. Epidemiology of haemoglobin disorders in Europe: An overview. Scand J Clin Lab Invest. 2007;67(1): Henderson S, Timbs A, McCarthy J, et al. Incidence of haemoglobinopathies in various populations the impact of immigration. Clin Biochem. 2009;42(18): Makhoul NJ, Wells RS, Kaspar H, et al. Genetic heterogeneity of b thalassemia in Lebanon reflects historic and recent population migration. Ann Hum Genet. 2005;69(Pt 1): Villegas A, Ropero P, Gonzalez FA, et al. The thalassemia syndromes: Molecular characterization in the Spanish population. Hemoglobin. 2001;25(3): Kountouris P, Lederer CW, Fanis P, et al. IthaGenes: An interactive database for haemoglobin variations and epidemiology. PLoS One. 2014;9(7):e Bennani C, Bouhass R, Perrin-Pecontal P, et al. Anthropological approach to the heterogeneity of b-thalassemia mutations in northern Africa. Hum Biol. 1994;66(3): Pacheco P, Loureiro P, Faustino P, Lavinha J. Haplotypic heterogeneity of b-thalassaemia IVS I-1 (G!A) mutation in southern Portugal. Br J Haematol. 1996;94(4): Antonarakis SE, Kazazian HH, Orkin SH. DNA polymorphism and molecular pathology of the human globin gene clusters. Hum Genet. 1985;69(1): Samara M, Chiotoglou I, Kalamaras A, et al. Large-scale population genetic analysis for hemoglobinopathies reveals different mutation spectra in central Greece compared to the rest of the country. Am J Hematol. 2007;82(7): Flatz G, Wilke K, Syagailo YV, et al. b-thalassemia in the German population: Mediterranean, Asian and novel mutations. Hum Mutat. 1999;13(3): Aguilar Martinez P, Angastiniotis M, Eleftheriou A, et al. Haemoglobinopathies in Europe: Health & migration policy perspectives. Orphanet J Rare Dis. 2014;9:97.
131 DOI: / Thalassemia in the Czech and Slovak Populations 7 Downloaded by [Knihovna Univerzity Palackeho], [${individualuser.displayname}] at 06:40 10 March Thein SL. Molecular genetics of b thalassaemia. In: McArthur JR, Lee SH, Wong JEL, Ong YW, Eds. Education Programme of the 26th Congress of the International Society of Haematology, Singapore, August 25-29, Singapore: International Society of Haematology. 1996: Thein SL. Genetic insights into the clinical diversity of b thalassaemia. Br J Haematol. 2004;124(3): Sollaino MC, Paglietti ME, Perseu L, et al. Association of a globin gene quadruplication and heterozygous b thalassemia in patients with thalassemia intermedia. Haematologica. 2009;94(10): Schwartz E, Goltsov AA, Kaboev OK, et al. A novel frameshift mutation causing b-thalassaemia in Azerbaijan. Nucleic Acids Res. 1989;17(10): Solovyev GY, Goltsov AA, Surin VL, et al. Molecular nature of mutations causing b 0 -thalassaemia in Azerbaijan. Biomed Sci. 1990;1(3): Çürük MA, Yüreğir GT, Asadov CD, et al. Molecular characterization of b-thalassemia in Azerbaijan. Hum Genet. 1992; 90(4): Fei Y-J, Stoming TA, Kutlar A, et al. One form of inclusion body b-thalassemia is due to a GAA!TAA mutation at codon 121 of the b chain. Blood. 1989;73(4): Thein SL, Hesketh C, Taylor P, et al. Molecular basis for dominantly inherited inclusion body b-thalassemia. Proc Natl Acad Sci USA. 1990;87(10): Yamamoto Ku, Yamamoto Ki, Hattori Y, et al. Two b-thalassemia mutations in Japan: Codon 121 (GAA!TAA) and IVS-I-130 (G!C). Hemoglobin. 1992;16(4): Giordano PC, Harteveld CL, Michiels JJ, et al. Phenotype variability of the dominant b-thalassemia induced in four Dutch Supplementary material available online families by the rare cd121 (G!T) mutation. Ann Hematol. 1998; 77(6): Cao A, Galanello R. b-thalassemia. Genet Med Off J Am Coll Med Genet. 2010;12(2): Rivella S. b-thalassemias: Paradigmatic diseases for scientific discoveries and development of innovative therapies. Haematologica. 2015;100(4): Divoky V, Svobodova M, Indrak K, et al. Hb Hradec Kralove (Hb HK) or a 2 b 2 115(G17)Ala!Asp, a severely unstable hemoglobin variant resulting in a dominant b-thalassemia trait in a Czech family. Hemoglobin. 1993;17(4): Popovich BW, Rosenblatt DS, Kendall AG, Nishioka Y. Molecular characterization of an atypical b-thalassemia caused by a large deletion in the 5 0 b-globin gene region. Am J Hum Genet. 1986; 39(6): Waye JS, Eng B, Patterson M, et al. Novel b-thalassemia mutation in a b-thalassemia intermedia patient [poly A (AATAAA! GATAAA)]. Hemoglobin. 2001;25(1): Thein SL. The molecular basis of b thalassemia. Cold Spring Harb Perspect Med. 2013;3(5):a Gallienne AE, Dreau HMP, Schuh A, et al. Ten novel mutations in the erythroid transcription factor KLF1 gene associated with increased fetal hemoglobin levels in adults. Haematologica. 2012; 97(3): Sutton M, Bouhassira EE, Nagel RL. Polymerase chain reaction amplification applied to the determination of b-like globin gene cluster haplotypes. Am J Hematol. 1989;32(1): Pritchard CC, Tait JF, Buller-Burckle AM, Mikula M. Annotation error of a common b 0 -thalassemia mutation (619 bp-deletion) has implications for molecular diagnosis. Am J Hematol. 2010; 85(12):978.
132 Příloha 4: Talasemie (domácí publikace bez IF). Divoká M., Partschová M., Pospíšilová D., Orviská M., Lapčíková A., Indrák K., Čermák J., Divoký V. Alfa-talasemie u 45 českých rodin a 37 rodin cizinců žijících v České republice. Přehled literatury a molekulárně-genetická diagnostika. Trans Hemat Dnes. 22, 2016, No 3, p
133 Alfa-talasemie u 45 českých rodin a 37 rodin cizinců žijících v České republice: přehled literatury a molekulárně-genetická diagnostika Divoká M. 1, Partschová M. 2, Pospíšilová D. 3, Orviská M. 1, Lapčíková A. 1, Indrák K. 1, Čermák J. 4, Divoký V. 1, 2 1 Hemato-onkologická klinika, Léka ská fakulta Univerzity Palackého a Fakultní nemocnice Olomouc 2 Ústav biologie, Léka ská fakulta Univerzity Palackého v Olomouci 3 Dětská klinika, Léka ská fakulta Univerzity Palackého v Olomouci a Fakultní nemocnice Olomouc 4 Ústav hematologie a krevní transfuze, Praha Souhrn Alfa-talasemie jsou nejrozší enější monogenně dědičná onemocnění na světě. Jedná se o vrozené anémie s autozomálně recesivní dědičností charakterizované kvantitativní redukcí syntézy α-globinových etězců molekuly hemoglobinu. Společně s dalšími typy talasemií a se strukturními hemoglobinovými variantami jsou azeny do skupiny chorob s poruchou tvorby nebo struktury globinových etězců - hemoglobinopatií. Výskyt většiny hemoglobinopatií je soust eděn p edevším do malarických oblastí Protože globinové mutace poskytují heterozygotním nosičům ochranu p ed malárií, došlo v průběhu evoluce pravděpodobně k selektivnímu zvýhodnění mutantních alel globinových genů v oblastech s vysokým výskytem malárie. Alfa-talasemie jsou nejčastěji způsobeny delecemi úseků DNA v α-globinovém lokusu na 16. chromozomu a vyznačují se mikrocytární hypochromní anemií s různými fenotypovými stupni závažnosti, od témě asymptomatických forem až po život ohrožující hemolytické anémie. Jsou rozší eny zvláště v jihovýchodní Asii, ve St edomo í, v Africe a Indii, ve st ední Evropě jsou vzácné. P esto i v našich zeměpisných ší kách je na p ítomnost α-talasemie nutné myslet jako na možnou p íčinu vrozené mikrocytární anémie. Pro správnou diagnostiku α- talasemií je nutný soubor hematologických, biochemických a molekulárně-genetických vyšet ení. Cílem tohoto sdělení je upozornit na variabilní fenotyp a gen otyp těchto onemocnění v České republice u českých rodin i rodin cizinců žijících v ČR. Klíčová slova hemoglobinopatie, α-talasemie, onemocnění HbH, molekulárně-genetická diagnostika
134 Summary Divoká M., Partschová M., Pospíšilová D., Orviská M. 1, Lapčíková A. 1, Indrák K., Čermák J., Divoký V. Alpha-thalassemia in 45 Czech and 37 immigrant families: review of the literature and molecular diagnosis Alpha-thalassemia is the most common monogenic gene disorder in the world. It is an inherited autosomal recessive disorder characterized by reduced production of the α-globin chains of hemoglobin. It belongs, together with other types of thalassemias and hemoglobinopathies, to common globin gene disorders widely distributed due to their natural selection through protection of carriers against severe malaria. Alpha-thalassemia most frequently results from deletions removing segments of DNA from the α-globin gene cluster on chromosome 16 and is characterized by a microcytic hypochromic anemia, with a clinical phenotype varying from almost asymptomatic to a lethal hemolytic anemia. Alphathalassemia is especially frequent in populations originating from the South East Asia, Mediterranean region, Africa and Indian subcontinent, with rare occurrence in Central Europe. However, also in our region the presence of α-thalassemia should be considered as a possible cause of hereditary microcytic anemia. Specialized diagnostic approaches that are needed for correct diagnosis of α-thalassemia involve molecular analysis required to confirm the hematological and biochemical observations. The aim of this study is to report the variable phenotype and genotype of these disease states in the Czech families and in immigrants living in the Czech Republic. Key words: hemoglobinopathy, α-thalassemia, HbH disease, molecular diagnostics
135 Úvod Alfa-talasemie je nejrozší enější monogenně dědičné onemocnění na světě, asi 5 % světové populace nese některou z α-talasemických alel (1). Alfa-talasemie, spolu s dalšími typy talasemií a s endemickými hemoglobinovými variantami byly selektovány v průběhu evoluce v malarických oblastech, neboť heterozygotům p inášejí výhodu v protekci proti malarickému parazitu Plasmodium falciparum (2). Talasemie i hemoglobinové varianty se mohou ovšem vyskytnout v každé etnické skupině a vzhledem ke vzrůstající migraci obyvatel se stále častěji rozši ují do oblastí st ední a severní Evropy, do severní Ameriky a také Austrálie. Právě ve st ední Evropě bylo d íve toto onemocnění léka i opomíjeno. Díky historické i současné imigraci se talasemie a hemoglobinové varianty objevují stále častěji i v naší populaci. V České republice a na Slovensku jsou talasemie nejčastější p íčinou vrozené mikrocytární anemie (3), ale pozornost byla dosud věnována témě výhradně -talasemiím (3-6). Alfa-talasemie byly dosud v ČR charakterizovány na molekulární úrovni jen výjimečně (3, 4); v jednom p ípadě se jednalo o nový unikátní molekulární mechanizmus vedoucí k α-talasemii (7, 8). V tomto sdělení p inášíme p ehled literatury a vlastní zkušenosti s diagnostikou α-talasemických alel. Uvádíme variabilní fenotyp a molekulárně-genetickou analýzu těchto onemocnění u 45 rodin českého původu a 37 rodin cizinců žijících v České republice. Genotypová a fenotypová variabilita α-talasemií Poměr α a -globinových etězců v molekule hemoglobinu (Hb) je fyziologicky jedna ku jedné. Nerovnováha vzájemného poměru globinových etězců je charakteristická pro talasemie; α-talasemie se vyznačují redukovanou produkcí α-globinových etězců. Alfatalasemie jsou většinou způsobeny delecí funkčních α-globinových genů, méně často jsou α- globinové geny inaktivovány bodovou mutací. Haploidní lidský genom má dva α-globinové geny (obr. 1A), to znamená, že normální diploidní genotyp je αα/αα. Normální sada čty funkčních α-globinových genů může být snížena o 1, 2, 3 nebo o všechny 4 kopie genu a z toho následně vyplývají rozdílné klinické projevy a zvyšující se závažnost onemocnění (obr. 1B). Míru snížené produkce α-globinových etězců z lokusu vyjad uje označení α + -talasemie a α 0 -talasemie. U α + -talasemie je jen jeden ze dvou α-globinových genů na chromozomu deletován ( α/αα) nebo inaktivován bodovou mutací (α ND /αα, kde ND značí nondeletional ; d íve α T /αα). U α 0 -talasemie jsou oba α-globinové geny na chromozomu inaktivovány delecí nebo bodovou mutací. Stupeň mikrocytózy, hypochromázie a anemie závisí na počtu
136 mutovaných α-globinových genů a dob e koreluje s mírou redukce syntézy α-globinových etězců (1). Obr. 1. Struktura α-globinového lokusu na chromozomu 16 a genotypové varianty delečních α-talasemií. A) Rodina (klastr) α-globinových genů leží v lokusu v blízkosti telomery na chromozomu 16. Klastr je dlouhý 29 kb a obsahuje embryonální gen ζ a dva α-globinové geny fetálního a dospělého typu α2 neboli HBA2 a α1 neboli HBA1 (plné boxy), které jsou uspo ádány na chromozomu ve směru jejich exprese během ontogeneze. Exprese těchto genů je ízena erytroidně-specifickou regulační oblastí, která leží 40 kb proti směru exprese ζ- globinového genu a nazývá se pozitivní regulační oblast HS-40. Prázdné boxy značí nefunkční kopie genů pseudogeny. Alfa-globinový lokus se nachází v transkripčněpermisivní chromatinové oblasti, to znamená, že se nachází v trvale otev ené chromatinové doméně a HS-40 oblast působí jako zesilovač globinové exprese. B) Naho e vlevo: normální diploidní genotyp. Naho e vpravo: heterozygot pro α + -talasemii, tzv. němé (nebo tiché) nosičství. V krevním obraze a v poměru syntézy α : globinových etězců se prakticky neprojevuje a prokazujeme je jen p i genetické analýze. Uprost ed: u homozygotů pro α + -
137 talasemii (nejčastěji delece dvou genů, α/ α) nebo u heterozygotů pro α 0 -talasemii ( /αα) hovo íme o nosičství α-talasemie. Projevuje se mírnou mikrocytózou, zvýšeným počtem erytrocytů a nerovnováhou v syntéze globinových etězců. Dole vlevo: ztráta t í α- globinových genů (dvojití heterozygoti pro α + -talasemii a α 0 -talasemii, nejčastěji / α), vede k chorobě HbH se st edně těžkou chudokrevností a s produkcí HbH ( 4). P i inkubaci s briliant-kresylovou mod í nacházíme v erytrocytech precipitovaný HbH. Dole vpravo: chybění čty α-globinových genů, tj. homozygotní stav pro α 0 -talasemii ( / ), není slučitelné s životem a vede ke vzniku hydrops fetalis nebo syndromu Hb Bart s ( 4). Rodí se hydropicky změněné dítě s ascitem, velkými játry, erytroblastózou, retikulocytózou, terčovitými buňkami s p evažujícím Hb Bart s a malým podílem HbH a Hb Portland (ζ2 2). Upraveno podle Divoký, 2013 (9). Klinické formy α-talasemií Název α-talasemie obecně zahrnuje všechna onemocnění s deficitem tvorby α- globinových etězců, které jsou součástí tetrametru Hb molekuly. Všechny lidské Hb molekuly jsou tetramery, sestávající ze dvou různých párů globinových etězců. Jeden pár globinových etězců je produkován geny α-globinového lokusu (ve fetálním období i v dospělosti jsou to α2 neboli HBA2 a α1 neboli HBA1 geny, jejichž kódující sekvence jsou identické). Druhý pár etězců je produkován geny -globinového lokusu (ve fetálním období jsou to G neboli HBG2 a A neboli HBG1 geny, v dospělosti neboli HBB gen). Snížená produkce α-globinových etězců vede ve fetálním období k nadbytku -globinových etězců, které se formují do 4 tetramerů, a vzniká tak Hb Bart s, v období dospělosti k nadbytku globinových etězců, které se formují do 4 tetramerů za vzniku nestabilního HbH. Na základě fenotypu rozlišujeme u nosičství pro α-talasemii dva typy: tiché nosičství s inaktivací (většinou delecí) jednoho α-globinového genu ( α/αα), které se v krevním obraze témě neprojevuje a prokázat jej lze jen pomocí molekulárně genetického vyšet ení. Výskyt tohoto genotypu je v České republice častý. U druhého typu nazývaného nosičství α-talasemie se jedná nejčastěji o deleci dvou α-globinových genů. adíme sem tedy buď homozygoty pro α + -talasemii ( α/ α) nebo heterozygoty pro α 0 -talasemii ( /αα) (viz obr. 1B). Nosičství α- talasemie se projevuje mírnou mikrocytární anemií se zvýšeným počtem erytrocytů. Výskyt těchto jedinců je v České republice relativně vzácný. HbH onemocněním (někdy označovaným jako choroba HbH) trpí jedinci se ztrátou t í α-globinových genů, v p ípadě deleční formy α-talasemie se jedná o jedince s ( / α)
138 genotypem, jde tedy o složené heterozygoty pro α + -talasemii a α 0 -talasemii. Tito nemocní většinou produkují méně než 30 % normální hladiny α-globinu, trpí st edně těžkou anemií (Hb mezi g/l) s různým zastoupením HbH (0,8 40 %), mají splenomegalii (která může být závažná). Samotný HbH má vysokou afinitu ke kyslíku a je nestabilní (10; obr. 1B). Choroba HbH výrazně neovlivňuje kvalitu života; v České republice se vyskytuje ojediněle u imigrantů resp. jejich potomků (viz dále). Hb Bart s ( 4) syndrom nebo hydrops fetalis je stav neslučitelný se životem, jedná se o homozygotní stav pro α 0 -talasemii, tedy o chybění všech čty α-globinových genů ( / ). Většinou se rodí novorozenci s hydropsem a ascitem, velkými játry, erytroblastózou a retikulocytózou. Nejvíce hemoglobinu v erytrocytech u novorozenců s Hb Bart s hydrops fetalis syndromem je tvo eno fyziologicky nefunkčními homotetramery 4 a 4. Mají také proměnlivé množství embryonálního Hb Portland, což je u těchto dětí jediný funkční hemoglobin a jediný p enašeč kyslíku, který je drží p i životě do narození (11). Vzácně jsou známé p ípady dlouhých delecí, které postihují kromě α-globinových genů také další geny v jejich okolí. Toto onemocnění je asociované s poruchami vývoje (zahrnující také duševní poruchy) a je označováno jako α-talasemie/syndrom mentální retardace asociovaný s chromozomem 16 (ATR-16 syndrom; 12). Byly popsány také p ípady vzácné formy mentální retardace spojené s α-talasemií, způsobené abnormalitami ATRX genu ležícím na chromosomu X (ATR-X syndrom). U těchto nemocných je duševní porucha více závažná a dysmorfické změny jsou nápadně podobné mezi jednotlivými pacienty. Gen ATRX kóduje protein ze skupiny helikáz/atpáz, který ovlivňuje chromatinovou strukturu a transkripci různých genů, mezi nimi i α-globinových genů. Mutace ATRX vedou ke globálnímu potlačení exprese těchto genů, mezi nimi i genů důležitých pro normální rozvoj CNS. U nás (v ČR nebo na Slovensku) jsme dosud ATR syndrom nediagnostikovali. Vzácné jsou získané formy α-talasemie, asociované s myelodysplastickým syndromem (syndrom ATMDS). Ve většině p ípadů je ATMDS syndrom asociován se získanými mutacemi v ATRX genu (12). Molekulární podstata α-talasemií Deleční α + -talasemie Vzhledem k tomu, že dva α-globinové geny v lokusu vznikly v evoluci duplikací, vyznačuje se tato oblast DNA několika homologiemi, které mohou vést k nerovnoměrným reciprokým rekombinacím (interchromozomálně, méně často intrachromozomálně). Výsledkem je vznik alel s jedním funkčním α-globinovým genem a alel s triplikacemi α-
139 globinových genů (13). Vzniká tak velmi častá 3.7 kb dlouhá α-talasemická delece, produkující chromozom pouze s jedním funkčním α-genem ( α 3.7 ) (obr. 2) a α-triplikovaná alela (ααα anti3.7 ) bez talasemického efektu. Stejně tak reciproká rekombinace v průběhu meiózy mezi homologními segmenty DNA způsobuje běžnou 4.2 kb dlouhou deleci ( α 4.2 ) (obr. 2) a ααα anti4.2 chromozom (13). Další p ípady rekombinací mezi takto vzniklými chromozomy (α, αα a ααα) pak mohou způsobit kvadruplikaci α genů (αααα) nebo kvintuplikaci (ααααα) a další p eskupení (14). Na rozdíl od delecí vedoucích k talasemickému fenotypu se samotné zmnožení α-globinových genů u jinak zdravých jedinců klinicky většinou neprojevuje. Problém ovšem nastává u dvojitých heterozygotů pro alelu se zmnoženými α-globinovými geny a -talasemickou alelu - dochází tak ke zvýšení nerovnováhy α a etězců a ke zvýraznění chudokrevnosti, což bylo vzácně potvrzeno i u nemocných v ČR (6, 15). Deleční α 0 -talasemie V současné době je známo asi 50 typů delecí HBA lokusu, které buď kompletně nebo částečně deletují oba α-globinové geny s následným zastavením syntézy α-globinových etězců in vivo (13). Nejčastější jsou delece MED rozší ená mezi populacemi ve St edomo í a SEA vyskytující se p evážně v jihovýchodní Asii (1; obr 2). Homozygoti pro tento typ delecí trpí Hb Bart s hydrops fetalis syndromem. Složení heterozygoti pro tento typ delece a delece pro α + -talasemii, která způsobí chybění jednoho α-globinového genu, mají HbH onemocnění. Tyto zatím zmíněné typy delecí postihují pouze HBA lokus a vyskytují se i v ČR (viz níže). Jiné delece postihují i jiné p iléhající geny, což nemusí mít výrazný vliv na zhoršení fenotypu. Některé známé delece odstraňují i ζ-gen současně s α-geny, a i když heterozygoti vykazují normální vývoj, u homozygotů je nepravděpodobné p ežití období časného stádia gravidity vzhledem k tomu, že ani embryonální (ζ 2 2 ) a ani fetální (α 2 2 ) hemoglobiny nemohou být syntetizovány (13). Tyto typy delecí byly zjištěny i v české populaci (viz níže). Vzácné delece vedoucí k α 0 -talasemii odebírají regulační oblast HS-40, která se nachází 40 kb 5 od ζ-globinového genu (obr. 1A), p ičemž zanechávají α-globinové geny intaktní. HS-40 je esenciální pro α-globinovou expresi, delece této oblasti vede k úplné ztrátě exprese α-globinových genů v poloze cis. Fenotyp je tedy shodný jako u vzácných delecí celého HBA lokusu. Tyto delece se sporadicky vyskytují v různých populacích (10), vzácně i u českých rodin (viz níže, obr. 2, tab. 1).
140 Vznik α-talasemie způsobené zastavením transkripce neaktivním chromatinem u polské rodiny žijící v České republice byl popsán jako nový mechanizmus vzniku tohoto onemocnění (16). Delece dlouhá 18 kb zahrnuje α1 gen a p ilehlou 3 oblast a způsobí utlumení exprese α2 globinového genu v poloze cis. Díky deleci se do lokusu p emístí represivní chromatin, který zastaví α2 transkripci (7, 8). Heterozygoti pro tuto mutaci trpí mírnou hypochromní mikrocytární anemií, mají snížený poměr syntézy α/ globinových etězců a HbH inkluze v erytrocytech. Obr. 2. Schematické znázornění delecí HBA lokusu zjištěných v České republice u českých rodin a u rodin cizinců. Delece MED, delece celého lokusu a různě dlouhé delece regulační oblasti HS-40 jsou u nás popsány poprvé v tomto sdělení. Vysvětlení genů a pseudogenů v lokusu viz obr. 1A. HVR značí tzv. hypervariabilní oblasti v HBA lokusu. Vertikální šipky nad lokusem značí polohy MLPA prób použitých pro mapování delecí (viz Diagnóza a diagnostické metody). Nedeleční formy α-talasemie ( ND ) Alfa-talasemie jsou častěji způsobeny genovými delecemi, bodové mutace (jednonukleotidové záměny) vedoucí k α-talasemii jsou vzácnější (10, 13). Alfa-globinové geny mutované v kanonických sekvencích HBA1 a HBA2 ovlivňujících α-globinovou genovou expresi se označují jako (α ND nebo d íve α T ); bodová talasemická mutace může postihnout α2 (α ND α) nebo α1 (αα ND ) globinový gen. Většina těchto mutací ovlivňuje úpravu
141 (p epis a post-transkripční zpracování) mrna, mrna translaci nebo α-globinovou stabilitu. Bodové mutace mohou vést k částečnému (α + ) nebo úplnému (α 0 ) utlumení genové exprese, resp. produkce funkčních α-globinových etězců. Je známo několik desítek bodových mutací způsobujících α-talasemii; mnohé z nich jsou běžné i ve St edomo í a na Blízkém východě a lze je tedy očekávat i české a slovenské populaci. Aktualizovaný seznam těchto mutací je dostupný v databázi HbVar, Některé bodové mutace v terminačním kodonu vedou k prodlouženým Hb variantám s talasemickým fenotypem, jako nap. Hb Constant Spring, nejběžnější nedeleční α-talasemická mutace častá zvláště v některých oblastech jihovýchodní Asie. Jiné mutace způsobují vysoce nestabilní α-globinové varianty, nap. Hb Adana (10). Nedávno byla popsána odchylka jednoho nukleotidu v negenové oblasti globinového klastru, která vede k novému molekulárnímu mechanizmu vzniku α-talasemie (17). Jedná se o záměnu v sekvenci regulačního jednonukleotidového polymorfizmu (SNP, Single Nucleotide Polymorphism ), kterou se vytvá í nový transkripčně aktivní promotor v normálně transkripčně neaktivní oblasti ψζ1. Tato abnormální transkripční aktivace interferuje a zeslabuje expresi α-globinových genů. Diagnóza a diagnostické metody Iniciální laboratorní testování by mělo zahrnovat kompletní krevní obraz s popisem červených krvinek, bioanalytické testy zahrnující mě ení hladiny HbA 2, HbF a elektroforézu Hb. D íve bylo pro potvrzení α-talasemie nezbytné mě ení poměru syntézy α/ globinových etězců radioaktivně značeným leucinem; redukovaný poměr na méně než 0.8 svědčil pro α- talasemii (18). V současné době p evažují molekulární analýzy, které potvrdí podez ení na α- talasemii. Elektroforéza Hb slouží k separaci abnormálních hemoglobinových frakcí, je důležitá mimo jiné pro detekci Hb variant s α-talasemickým fenotypem. Precipitovaný HbH je v erytrocytech prokazován inkubací s brilliant-kresylovou mod í (obr. 3A). Molekulární analýza Molekulární diagnostice napomáhá etnický původ vyšet ovaných jedinců; rozdílné spektrum α-talasemických mutací bývá nalézáno v rozdílných populacích. D íve byla většina p eskupení HBA genů v rámci HBA lokusu charakterizována pomocí Southern blotu (obr. 3B). V současné době existují rychlé skríningové metody pro detekci delecí a jiných změn v HBA lokusu. Pro detekci sedmi nejčastějších α-talasemických delecí bylo vytvo eno multiplexní PCR (19; obr. 3C). Tato metoda se používá pro určení dvou nejčastějších α + -talasemických delecí ( α 3.7 ; α 4.2 ) a dalších 5 α 0 -talasemických delecí, mezi nimi MED a SEA. Novější MLPA
.")
142 (Multiplex Ligation-dependent Probe Amplification) je založena na ligaci více párů prób (obvykle dlouhých), které hybridizují nap íč k analyzované oblasti. Následuje semikvantitativní amplifikace s použitím univerzálních tag PCR primerů a poté fragmentační analýza (obr. 3D). Tato metoda je hodnotnou alternativou k Southern blotu a doplňkovou k multiplexní PCR p i zjišťování jak známých tak neznámých delecí způsobujících α-talasemii (20). V p ípadě podez ení na bodovou mutaci (nedeleční mutaci) se rutinně používá sekvenování HBA1 a HBA2 genů.
143 Obr. 3. Diagnostika α-talasemických delecí. A) Precipitovaný HbH lze v erytrocytech prokázat inkubací s brilliant-kresylovou mod í, po obarvení jsou patrné inkluze tvo ené shlukem volných -globinových etězců v erytrocytu. B) Výsledek Southern blot analýzy prokazující dvě nejčastější α + -talasemické delece ( α 3.7 ; α 4.2 ) a triplikaci α-globinových genů. C) Výsledek multiplexního PCR v agarózovém gelu prokazující dvě nejčastější α + - talasemické delece ( α 3.7 ; α 4.2 ) a α 0 -talasemickou deleci MED.
144 D) Výsledek MLPA prokazující deleci regulační oblasti HS-40 a α + -talasemickou deleci α 3.7, které u složeného heterozygota vedou k chorobě HbH. Další informace k dané technologii a způsobu analýzy výsledků jsou dostupné na produkt SALSA MLPA P140 HBA probemix. Diferenciální diagnostika V některých p ípadech mají nosiči α + -talasemie normální hematologické výsledky, speciálně nosiči α 3.7 delece a nosiči nedelečních mutací v α1 genu. Někte í jedinci mohou mít normocytózu nebo hraniční hypochromázii bez anemie. Tato skutečnost může být zjištěna pouze náhodně p i rutinní molekulární analýze pro záchyt hemoglobinopatií. P íležitostně, speciálně v zemích, kde je talasemie vzácná, mohou být nosiči α-talasemie nesprávně označení za pacienty s anemií z nedostatku železa, zvláště pokud není status železa u pacienta správně zhodnocen. Vždy je nutná molekulární analýza, zvláště u tichého nosičství nebo nosičství α-talasemie k potvrzení diagnózy, protože i nosiči α-talasemické alely můžou vykazovat vyšší riziko p etížení organizmu železem (21). Výskyt α-talasemií v České republice přehled literárních údajů Alfa-talasemie byly považovány v České republice za velmi vzácné; jen několik p ípadů bylo charakterizováno na molekulární úrovni tradiční metodou Southernova p enosu a hybridizace (3, 4). Kromě α 3.7 delece zjištěné u několika jedinců byl u jedné rodiny polského původu žijící v ČR popsán již zmíněný nový typ unikátní delece 18 kb DNA postihující α1 gen a p ilehlou 3'-oblast, která vedla p ekvapivě k utlumení exprese α2 globinového genu v poloze cis, tj. k fenotypu, který je asociován s α-tal-1 genotypem (16). Bylo zjištěno, že 18 kb delece p emístila do klastru represivní chromatin, který spolu s protismyslnými transkripty p ilehlého genu (gen, jenž je p episován proti směru exprese α- globinových genů) zastaví α2 transkripci (7, 8). Vzácně byly také detekovány triplikace α- globinových genů (3, 4), někdy vedoucí ke zhoršení klinického projevu -talasemie (22). Diagnostika α-talasemií v České republice nové výsledky V průběhu posledních 10 let došlo k významnému rozší ení spektra α-talasemických mutací i počtu molekulárně-geneticky diagnostikovaných jedinců v ČR. Ke zp esnění
145 diagnostiky p ispělo využití nových molekulárně-genetických p ístupů multiplexní PCR a MLPA pro diagnostiku delečních forem α-talasemií a sekvenační analýzy HBA genů pro nedeleční formy α-talasemií. V souboru 82 rodin (98 členů) s podez ením na α-talasemii jsme odhalili u 78 rodin (92 jedinců) různé delece v rámci HBA lokusu; 45 rodin (53 jedinců) bylo českých, bez p edků z malarických oblastí nejméně ve t ech generacích rodu, ostatní byli cizinci žijící v ČR (tab. 1). U dvou jiných českých rodin (4 jedinců) jsme odhalili multiplikaci α-globinových genů (tab. 1) a u dalších dvou imigrantů jsme odhalili nedeleční α- talasemickou mutaci. Většina diagnostikovaných jedinců měla mírnou mikrocytární hypochromní anemii, s normálními hladinami HbA 2 a HbF, se standardní elektroforézou Hb a mírně zvýšeným počtem erytrocytů. Pro možnou p ítomnost α-talasemické alely svědčil rodinný výskyt fenotypu, v p ípadě rodin cizinců i etnický původ. Ve většině p ípadů se jednalo o tiché nosičství nebo nosičství α-talasemie, s běžnými typy delecí jednoho nebo dvou α-globinových genů, které jsou známé ze St edozemí nebo z jihovýchodní Asie (obr. 2). Rozsáhlé delece celého HBA lokusu, včetně regulační oblasti HS-40, byly detekovány u 6 českých rodin (11 členů) s klinickými projevy odpovídajícími nosičství α-talasemie (jednalo se vesměs o heterozygoty, tab. 1). U 4 rodin (z nich 3 českého původu) jsme detekovali různě dlouhé delece regulační oblasti HS-40, ale s intaktními α-globinovými geny (viz obr. 2), s fenotypem odpovídajícím ztrátě dvou α-globinových genů (nosičství α-talasemie) u 5 jedinců, a u 1 pacientky jedné rodiny s klinickými projevy odpovídajícími ztrátě t í α- globinových genů (choroba HbH). Molekulární analýzou této pacientky s italskými p edky jsme zjistili, že fenotyp HbH onemocnění je u ní způsoben inaktivací dvou α-globinových genů zmíněnou delecí HS-40 na jednom chromozomu v kombinaci s delecí jednoho α- globinového genu ( α 3.7 ) na druhém chromozomu. Stejně závažný fenotyp, tj. chorobu HbH, jsme detekovali u imigranta ze severní Afriky, u kterého byl p íčinný genotyp kombinací dvou častých delecí ze St edozemí ( α 4.2 / MED ) (tab. 1).

.")
146 Tab. 1. P ehled delečních forem α-talasemií a multiplikací α-globinových genů zjištěných v průběhu posledních 10 let v České republice. Je uveden celkový počet rodin a jedinců, za lomítkem počet českých rodin a počet členů těchto rodin z celkového počtu diagnostikovaných rodin/jedinců. a ) 15 z těchto rodin (16 členů) bylo prokazatelně asijského původu; b ) pacient s italskými p edky; c ) imigrant ze severní Afriky žijící v ČR; d ) u všech členů rodiny byla kvadruplikace HBA2 genu asociována s 0 -talasemickou mutací, což vedlo k fenotypu talasemie intermedia u těchto nemocných (6). Nedeleční formy α-talasemie jsme zatím diagnostikovali jen u dvou imigrantů žijících v ČR, u obou se jednalo o mutaci HBA2:c.95+2_95+6delTGAGG, označovanou také jako α-thal-2 (- 5nt) (23), která se běžně vyskytuje ve St edomo í. Závěr První systematická molekulárně-genetická analýza α-talasemií v ČR provedená na selektovaném souboru 82 rodin (98 jedinců) odhalila novými diagnostickými p ístupy 92 jedinců ze 78 rodin s delecemi v rámci HBA lokusu; 45 rodin (53 jedinců) bylo prokazatelně českého původu. Další 2 české rodiny (4 členové) měli v genotypu multiplikaci α- globinových genů a u dalších dvou imigrantů byla odhalena nedeleční α-talasemická mutace. U většiny diagnostikovaných jedinců s α-talasemií minor (37 cizinců/35 Čechů), byly nalezeny delece a u 2 cizinců bodové mutace α-globinových genů běžné v malarických
147 oblastech. Vzácně jsme odhalili rozsáhlé delece celého HBA lokusu nebo delece regulační oblasti lokusu HS-40, ale s intaktními α-globinovými geny; tyto typy delecí byly častěji zjištěny u českých rodin než u imigrantů. Podobné typy delecí jsou velmi sporadicky detekovány v různých populacích, včetně původních obyvatel západní a severní Evropy. U 3 českých rodin nebylo možné deleci HBA klastru blíže specifikovat. Prioritně byl v ČR charakterizován genetický podklad dvou pacientů s onemocněním HbH, první měl italské p edky, druhý byl imigrant z Afriky. Heterogenita α-talasemií je v ČR z ejmě daleko větší než se dosud soudilo. Navíc, α-talasemické alely budou se stupňující se migrací v ČR postupně p ibývat, a s tím bude stoupat i nutnost jejich p esné diagnostiky založené na molekulárněgenetických analýzách. Podíl autorů na rukopisu DM napsání rukopisu, provádění analýz, interpretace výsledků, p íprava finální verze rukopisu PM provádění analýz, revize rukopisu PD léčba pacientů, p ipomínkování rukopisu OM analýzy pacientských vzorků LA analýzy pacientských vzorků IK léčba pacientů, p ipomínkování rukopisu ČJ léčba pacientů, p ipomínkování rukopisu DV p íprava finální verze rukopisu, interpretace výsledků Seznam použitých zkratek Hb hemoglobin
148 ND kb ATR SNP PCR MLPA nondeletional, nedeleční kilobáze α-thalassemia with mental retardation Single Nucleotide Polymorphism, jednonukleotidový polymorfizmus polymerázová etězová reakce Multiplex Ligation-dependent Probe Amplification Poděkování: Léka ům hematologických pracovišť v České republice a na Slovensku za poskytnutí pacientských vzorků a za spolupráci. Tato práce byla podpo ena granty IGA_LF_2016_001 a IGA_LF_2016_014 Literatura 1. Piel FB, Weatherall DJ. The α-thalassemias. N Engl J Med, 2014;371(20): Weatherall DJ, Williams TN, Allen SJ, O'Donnell A. The population genetics and dynamics of the thalassemias. Hematol Oncol Clin North Am, 2010;24(6): Indrák K, Divoký V, Brabec V, et al. Molekulárně genetická charakteristika α, a δ thalassémií u 139 heterozygotů z 56 nep íbuzných rodin českého a slovenského původu. Vnitř Lék, 1993;39(10): Indrak K, Brabec V, Indrakova J, et al. Molecular characterization of -thalassemia in Czechoslovakia. Hum Genet, 1992;88(4): Divoký V., Walczysková S, Pospíšilová D, et al. Některé vzácnější formy hereditárních anémií vyskytující se v dospělé populaci v ČR a SR - -talasemie a nestabilní hemoglobinové varianty. Vnitř Lék, 2005;51: Divoka M, Partschova M, Kucerova J, et al. Molecular Characterization of - Thalassemia in the Czech and Slovak Populations: Mediterranean, Asian and Unique Mutations. Hemoglobin, 2016;Mar 9:1-7. DOI: / Barbour VM, Tufarelli C, Sharpe JA, et al. α-thalassemia resulting from a negative chromosomal position effect. Blood, 2000;96(3): Tufarelli C, Stanley JA, Garrick D, et al. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet, 2003;34(2):
149 9. Divoký V, Indrák K, Mojzíková R. Hemoglobinopatie: talasémie a strukturální Hb varianty. In: Molekulární hematologie. Praha: Galén, 2013, s ISBN Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis, 2010;5:13. DOI: / Chui DH. Alpha-thalassemia: Hb H disease and Hb Barts hydrops fetalis. Ann N Y Acad Sci, 2005;1054: Higgs DR, Buckle VJ, Gibbons R, Steensma D. Unusual Types of α Thalassemia. In Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. (Eds). Disorders of hemoglobin: genetics, pathophysiology and clinical management, 2 nd Cambridge University Press, 2009, s edition. Cambridge: 13. Higgs DR. The molecular basis of α thalassemia. In Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. (Eds). Disorders of hemoglobin: genetics, pathophysiology and clinical management, 2nd edition. Cambridge: Cambridge University Press, 2009, s Lam KW, Jeffreys AJ. Processes of de novo duplication of human α-globin genes. Proc Natl Acad Sci U S A, 2007;104(26): Indrak K, Fei YJ, LI HW, et al. A Czechoslovakian teenager with Hb E- 0 -thalassemia [IVS-I-1 (G A)] complicated by the presence of an -globin gene triplication. Ann Hematol, 1991; 63(1): Indrak K, Gu YC, Novotny J, Huisman TH. A new alpha-thalassemia-2 deletion resulting in microcytosis and hypochromia and in vitro chain imbalance in the heterozygote. Am J Hematol, 1993;43(2): De Gobbi M, Viprakasit V, Hughes JR, et al. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science, 2006; 312(5777): Clegg JB, Weatherall DJ: Haemoglobin synthesis in alpha-thalassaemia (haemoglobin H disease). Nature, 1967; 215: Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-pcr screen for common deletional determinants of α-thalassemia. Blood, 2000; 95(1): Harteveld CL, Voskamp A, Phylipsen M, et al. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing alpha- and beta-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J Med Genet, 2005; 42(12):
150 21. Sulovska L, Holub D, Zidova Z, et al. Characterization of iron metabolism and erythropoiesis in erythrocyte membrane defects and thalassemia traits. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub DOI: /bp [Epub ahead of print] 22. Indrak K, Fei Y-J, Li H-W, et al. A Czechoslovakian teenager with Hb E- 0 - thalassemia [IVS-I-1 (G A)] complicated by the presence of an α-globin gene triplication. Ann Hematol, 1991;63(1): Orkin SH, Goff SC, Hechtman RL. Mutation in an intervening sequence splice junction in man. Proc Natl Acad Sci U S A, 1981;78(8): RNDr. Martina Divoká Hemato-onkologická klinika, Léka ská fakulta Univerzity Palackého a FN Olomouc Hněvotínská Olomouc martina.divoka@fnol.cz Čestné prohlášení autora Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve st etu zájmů a vznik ani publikace článku nebyly podpo eny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
151 Příloha 5: Talasemie (zahraniční a domácí konference). Divoka, M., Partschova, M., Mojzikova, R., Piterkova, L., Horvathova, M., Cermak, J., Pospisilova, D., Indrak, K., Divoky, V. Molecular characterization of beta-thalassemia and hemoglobin variants in the Czech and Slovak populations: an update. EHA, Londýn, UK, Haematol. Hematol J., vol 96 (Suppl. 2), p. 626 (1593). Publikovaný abstract. Divoka, M., Mojzikova, R., Piterkova, L., Pospisilova, P., Parstchova, M., Horvathova, M., Pospisilova, D., Striezencova Laluhova, Z., Cermak, J., Indrak, K., Divoky, V. Molecular characterization of hemoglobinopathies and red cell enzymopathies in the Czech and Slovak population: an update. ASH, San Diego, USA, Blood (ASH Annual Meeting Abstracts), 118(21), 2011, Online - publikovaný abstrakt. Divoká, M., Partschová, M., Mojzíková, R., Piterková, L., Horváthová, M., Koláčková, M., Kalandrová, E., Čermák, J., Pospíšilová, D., Fábryová, V., Indrák, K., Divoký, V. Molekulární charakterizace beta-talasemií a hemoglobinových variant v české a slovenské populaci. XXV. Olomoucké hematologické dny, Olomouc, Trans Hemat Dnes, 17, 2011, str. 24. Přednáška. Divoká, M., Koláčková, M., Lipert, J., Novosadová, A., Orviská, M., Lapčíková, A., Láníková, L., Partschová, M., Pospíšilová, D., Divoký, V., Indrák, K. Hemoglobinopatie v české a slovenské populaci. XXVII. Olomoucké hematologické dny s mezinárodní účastí , Olomouc. Sborník abstrakt, str (P43/2401). Poster.

152
153 16 th Congress of the European Hematology Association Background. Thalassaemia is a group of inherited disorders of haemoglobins characterised by a significant decrease in the rate of synthesis of one or more globin chains, which in beta thalassaemia will be the beta chain. In adults the haemoglobin A2 (HbA2) comprises 2-3.5% of total haemoglobin. In some patients the proportion of HbA2 is raised. This is diagnostic for beta thalassaemia trait, but can also be seen in some unstable haemoglobins. Mutations which cause decreased β-globin gene expression are classified as either β+ (reduced level of β-globin synthesis) or β0 (no β-globin synthesis). Aim In this study the HbA2 was quantified using two different methods, capillary zone electrophoresis (CE) and high-pressure liquid chromatography (HPLC). The results was correlated with the mutations found via β globin gene sequencing in order to establish whether or not quantitation of HbA2 by either method can be used to predict the β-globin mutation type (β+ or β0). Method The CE, HPLC and β globin gene sequencing were used to evaluate a total of 31 subjects with HbA2 > 3.5% (indicative of beta thalassemia trait). Results The HbA2 results produced by CE were statistically significantly lower than those obtained by HPLC when compared using a paired t-test (p= <0.001). Three of the thirty one patients were found not to have any common beta globin gene mutations, but were included as the HPLC showed an HbA2 percentage of 3.6, 3.7 and 3.9 respectively. Out of the 31 patients, 8 cases were diagnosed as having heterozygous β0 mutations including 4 patients with codons 41/42 (-TTCT) mutations, 2 patients with codons 8/9 (+G) mutations, 1 patient with a codon 15 (- T) mutation and 1 patient with a codon 39 (CAG-TAG) mutation. Twenty patients were found to be heterozygous for β+ mutations including 8 patients with IVS-1-5 (G-C) mutations, 6 patients with IVS (G- A) mutations, 3 patients with -29 (A-G) mutations, 2 patients with -88 (C-T) mutations and 1 patient with an IVS-I-6 (T-C) mutation. When compared using the students t-test there was no difference between the HbA2 results obtained using HPLC for the β+ and β0 mutations cohorts (p=0.06). The same comparison performed using the HbA2 results obtained using CE showed a statistically significant difference between the β+ and β0 mutation cohorts (p=0.02). The predictive values of the measurement of HbA2 in establishing whether a patient has a β+ or β0 mutation calculated using Receiver operating characteristic (ROC) curves are and for HPLC and CE respectively. Summary/Conclusion. From the results of this study the CE is more sensitive in distinguishing between β+ and β0 mutations. Although this difference is relatively modest it is suggestive of a trend which may be more profound if the cohort was larger. The HbA2 level was significantly lower with the CE analyser. No attempt was made to compare the different β+ and β0 mutations as the sample size was insufficient MOLECULAR CHARACTERIZATION OF BETA-THALASSEMIA AND HEMOGLOBIN VARIANTS IN THE CZECH AND SLOVAK POPULATIONS: AN UPDATE M Divoka, 1 M Partschova, 2 R Mojzikova, 2 L Piterkova, 2 M Horvathova, 2 J Cermak, 3 D Pospisilova, 1 K Indrak, 1 V Divoky 2 1Faculty Hospital, Olomouc, Czech Republic 2Faculty of Medicine, Palacky University, Olomouc, Czech Republic 3Institute of Hematology and Blood Transfusion, Prague, Czech Republic Background. β-thalassemia is considered to be a rare disorder in Middle Europe. Similarly to other non-malaria regions, the presence of β-thalassemia in Middle Europe reflects the historical as well as recent immigration and demographic changes that have influenced the genetic variability of the current populations living in this area. Aims. To assess the frequency and spectrum of mutations on the β-globin gene in Czech and Slovak patients with clinical symptoms of β-thalassemia or δβ-thalassemia. The results of the initial part of this research were published almost two decades ago (Indrak et al., Hum Genet 1992; 88: ) and the aim of this work was to update this original report. Patients and Methods: Nearly 380 cases from seven hematological centers of Czech and Slovak Republics were analyzed. Blood samples were available for blood cell count measurements, hemoglobin (Hb) electrophoresis, chromatography and for genetic analyses. Results. Twenty-two β-thalassemia mutations were identified in more than 260 heterozygotes from 152 unrelated families of Czech or Slovak descent. Most of the mutations were of Mediterranean origin and accounted for 70% of patients. Newly discovered insertion of transposable element L1 into the β-globin gene represents a novel etiology of β-thalassemia due to a silencing effect of repressive chromatin associated with retrotransposon insertion (Piterkova et al, Heamatologica 2011; 96(s2): 417-8, (EHA meeting abstract)). The list of abnormal hemoglobins now contains 14 β-globin variants, involving the rare high oxygen affinity Hb Olomouc associated with familiar polycythemia and two unique Heinz body hemolytic anemia variants (unstable Hb Hradec Kralove and Hb Hana). Conclusions. In the Czech and Slovak populations, β-thalassemia appears to be an uncommon disorder, which, however, must be considered as the prevailing cause of congenital hypochromic microcytic anemia. All but four studied patients were heterozygous carriers, manifesting thalassemia minor, with rare exceptions of dominantly inherited β-thalassemia with phenotype that ranged from severe thalassemia minor to thalassemia intermedia. Three of the four homozygous or double-heterozygous β- thalassemia patients were recent immigrants from malaria countries. Genetic drift and migration in the past as well as recent immigration are responsible for introduction of the Mediterranean alleles, while several mutations, described in single families, originated locally. Funding. Supported by grants NT11208 and NS /2009 (Ministry of Health Czech Republic); MSM (Ministry of Education, Youth and Sports) of the Czech Republic; student projects LF_2011_006 and LF_2011_011 of the Palacky University GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY A SINGLE INSTITUTION EXPERIENCE K Palla, E Perraki Chania General Hospital, Chania, Greece Background. Glucose-6-phosphate dehydrogenase deficiency(g-6pd) is an X-linked disorder affecting red cell metabolism. Its distribution varies significantly among different geographic regions.in Greece where this disease is endemic, an estimated males and females are affected. The main clinical manifestations are acute hemolytic anemia and jaundice triggered by infection or ingestion of fava beans or oxidative drugs. Aims. To present data indicating the frequency of G6PD deficiency in the population admitted to the hospital of Chania city (Crete island) where there is extensive consumption of fresh or dried fava beans and previously have been reported several cases of favism Methods: During 13 -year period ( ), tests for the quantitative measurement of G-6PD activity by enzymatic colorimetric assay by a commercial kit (Trinity biotech, Menarini) were conducted on 1397 samples.any individual with an activity below 4,6(U/g Hb) and 146 (U/1012 RBC) was considered G-6PD deficient. Results: Of the 1397 (1030 males, 367 females) screened, 267 (147 males, 120 females) were children. Among children 14/267 (5, 24%) were immigrants and 32/267 (11,98%)were found to have G-6PD deficiency. Complete enzyme deficiency was shown in 19/267(7,11%) males whereas 2/267 (0.74%) were immigrants. Moderate enzyme deficiency was identified in 13/267 (4, 86%)females. 4/267 (1, 49%) children were admitted to hospital with G6PD deficiency related acute hemolysis.the children with hemolysis were males, (2/5 were immigrants), younger than 5 years old and have consumed fava beans. Of the adults (220males, 910 females) 65/1130 (5,75% )were deficient in G6PD. Complete enzyme deficiency was shown in 40/1130 (3,53%) males and 6/1130(0.53%) were females. Moderate enzyme deficiency was identified in 19/1130 (1, 68%) females. The overall incidence of the deficiency in screened population (97/1397)was 6,94%. The rate among men was 59/1030 (5,73%) and among females, 38/367 (10,35%), with the male to female ratio 1:2. Conclusion: G6PD deficiency seems to affect more females than males. As neonatal screening for G6PD is long established in Greece,clinical cases of favism are observed rarely. Acute hemolysis was found only in young children. We believe that the screening with a comprehensive education program should be performed for young children in order to prevent the occurrence of hemolysis THE ROLE OF REGULATORY T CELLS AND FOXP3 EXPRESSION IN GREEK B-THALASSEMIA MAJOR PATIENTS E Farmaki, M Economou, E Vlachaki, V Tzimouli, A Kondou, A Taparkou, A Teli, V Perifanis, M Athanassiou-Metaxa Aristotle University of Thessaloniki,'Hippokration' General Hospital, Thessaloniki, Greece Background: The suppressive/immunomodulatory function of CD4+CD25+FoxP3+ regulatory T (Treg) cells is crucial for the maintenance of immune homeostasis. Although a large number of studies have been performed on immune status of patients with β-thalassemia major, including T and B-lymphocyte subpopulations, little information is available regarding the role of Treg cells in this patient group. Aim: The aim of the present study was to determine B cells and T cells subpopulations, 626 haematologica 2011; 96(s2)
154 Advertisement Molecular Characterization of Hemoglobinopathies and Red Cell Enzymopathies in the Czech and Slovak Populations: An Update Martina Divoka, Renata Mojzikova, Lucie Piterkova, Pavla Pospisilova, Martina Partschova, Monika Horvathova, Dagmar Pospisilova, Zuzana Laluhova Striezencova, Jaroslav Cermak, Karel Indrak, Vladimir Divoky Blood :5307; Article Info & Metrics e-letters Abstract Ab stract 5307 Background: Thalassemias are rare disorders in Middle Europe. However, as a result of historical and recent migration, thalassemias became common cause of congenital anemia in the Czech and Slovak populations. Abnormal hemoglobin variants and red-cell enzymopathies are rare cause of congenital anemia in this region. The aim of this work was to update the original reports of this research published almost two decades ago (Indrak et al., Hum Genet 1992; 88: , Xu et al., Blood 1995; 85:257 63, Lenzner et al., Blood 1997; 89:1793 9). We assessed the frequency and spectrum of -globin gene mutations in the patients with clinical symptoms of -thalassemia or δ, -thalassemia, the -globin gene status in the patients with clinical symptoms of -thalassemia, and we characterized red cell enzymopathies on molecular level in the Czech and Slovak populations. Patients and methods: Nearly 390 cases with clinical symptoms of thalassemia or hereditary nonspherocytic hemolytic anemia from several centers of Czech and Slovak Republic were analyzed. Hematological parameters, hemoglobin electrophoresis and enzyme activities were
155 measured by standard procedures. Genomic DNA was used for PCR-sequencing analysis. Results: We identified 22 -thalassemia mutations in more than 260 heterozygotes; most of the mutations were of Mediterranean origin. The newly discovered insertion of transposable element L1 into the HBB gene represents a novel etiology of -thalassemia due to a silencing effect of repressive chromatin associated with retrotransposon insertion. The list of abnormal hemoglobins now contains 14 -globin variants, involving Heinz body hemolytic anemia variant Hb Hana ( 63(E7) His-Asn), phenotype of which was worsened by concomitant partial glutathione reductase deficiency (Mojzikova et al., Blood Cells Mol Dis 2010; 45:219 22). Several G6PD and PK variants were described in the Czech and Slovak populations; the G6PD variants include G6PD Olomouc, G6PD Varnsdorf and G6PD Praha. Recently, we identified a new frameshift mutation c. 1553delG (p. Arg518fs) at the homozygous state in exon 11 of the PKLR gene of the pediatric patient who suffered from transfusion dependent hemolytic anemia with Hb=9.4 g/dl, Ret=4.5%. His red cells PK activity was 4.52 IU/gHb (normal range IU/gHb). The mutation occurs in C domain of PK-R subunit containing the binding site for fructose-1,6- bisphosphate. The patient's extremely elevated level of growth differentiation factor 15 (GDF15, 3577 pg/ml, healthy controls pg/ml) could explain hereditary hemochromatosis and signs of iron overload in this patient. C o nclusio ns: In the Czech and Slovak populations, hemoglobinopathies and red-cell enzymopathies appear to be an uncommon disorder, which, however, must be considered as the prevailing cause of congenital anemia. Most of the thalassemia patients were heterozygous, manifesting thalassemia minor. Most of the hemoglobin variants were described in single families, some of them originated locally. Among hemolytic anemias due to red-cell enzymopathies is the most frequent PK deficiency. This work was supported by grants NT11208, NS10281 (Ministry of Health Czech Republic), MSM (Ministry of Education, Youth and Sports) and student projects LF_2011_006 and LF_2011_011 of the Palacky University. Disclo sure s: No relevant conflicts of interest to declare by The American Society of Hematology Back to top Advertisement

156 ROČNÍK 17 ČERVEN / 2011 ČASOPIS SPOLEČNOSTI PRO TRANSFUZNÍ LÉKAŘSTVÍ A ČESKÉ HEMATOLOGICKÉ SPOLEČNOSTI XXV. Olomoucké hematologické dny s mezinárodní účastí XV. Konference ošetřovatelství a zdravotnických laborantů 4 rd Symposium on Advances in Molecular Hematology Olomouc, Supplementum VYDÁVÁ ČESKÁ LÉKAŘSKÁ SPOLEČNOST J. E. PURKYNĚ ISSN INDEXED IN EMBASE/Excerpta Medica Excerpováno v BIBLIOGRAPHIA MEDICA ČECHOSLOVACA Indexováno a excerpováno v databázi SCOPUS 2
157 Supplementum 2/2011 XXV. olomoucké hematologické dny tována rovněž etiologická souvislost infekce HP se sideropenickou anémií. V etiologii anémie mohou hrát roli ztráty krve, pokles ph žaludeční šťávy a spotřeba Fe pro metabolismus bakterií. Jednoznačná souvislost infekce HP se sideropenickou anémií u dětí však dosud nebyla prokázána. Soubor pacientů a metody. V letech bylo ve specializované hematologické ambulanci vyšetřeno 61 dětí se sideropenickou anémií. Jednalo se o 19 chlapců a 42 dívek ve věku 3-18 let. Diagnóza sideropenické anémie byla stanovena na podkladě detailní analýzy krevního obrazu a biochemických markerů metabolizmu železa. Po vyloučení gynekologické příčiny u dívek bylo provedeno vyšetření GIT včetně okutního krvácení a stanovení antigenu HP ve stolici. Výsledky Infekce HP byla prokázána u 33 (54%) pacientů (11 chlapců a 22 dívek) ve věku 5-18 let (medián 14 let). Průměrná hodnota Hb byla 83 g/l (rozptyl g/l). Hladina feritinu se pohybovala v rozmezí 0,9-18 ug/l. U všech pacientů byl pozitivní nález antigenu HP ve stolici, infekce byla potom verifikována při gastroskopickém vyšetření. 12 pacientů (14%) mělo klinické příznaky infekce v podobě bolestí břicha nebo pocitu plnosti v epigastriu. Pouze u 6 pacientů (8,2%) byl pozitivní nález okultního krvácení. U 31 pacientů došlo po léčbě infekce HP k normalizaci uvedených ukazatelů nejdéle za 3 měsíce od zahájení léčby, u dvou pacientek s refrakterní anemií jsme prokázali recidivu infekce HP s nutností opakované léčby. Závěr Rozdíl mezi výskytem infekce HP udávaným v dětské populaci (do 10%) a výskytem prokázané infekce v souboru pacientů se sideropenickou anémií (54%) je vysoce statisticky významný (p<0,00001), naše nálezy tedy prokázují infekci Helicobacterem pylori jako příčinu sideropenické anemie. Infekce Helicobacterem pylori by měla patřit do diferenciální diagnostiky sideropenické anémie především u dospívajících dívek. Včasným provedením gastroskopie můžeme tak předejít zdlouhavému neúspěšnému léčení preparáty Fe u těchto pacientů, u kterých by později tato diagnóza byla stanovena a onemocnění řádně léčeno. Podporováno výzkumným záměrem MSM a grantem LF Molekulární charakterizace beta-talasemií a hemoglobinových variant v české a slovenské populaci Divoká Martina, Partschová Martina, Mojzíková Renáta, Piterková Lucie, Horváthová Monika, Koláčková Monika, Kalandrová Eliška, Čermák Jaroslav, Pospíšilová Dagmar, Fábryová Viera, Indrák Karel, Divoký Vladimír (Hemato-onkologická klinika FN a LF UP, Olomouc; Ústav biologie LF UP, Olomouc; ÚHKT, Praha; Hematológia, Nemocnica sv. Michala, Bratislava SK) Úvod: Nejčastější příčinou vrozené mikrocytární anemie v České republice a na Slovensku jsou ß-talasemické alely a to i přesto, že jejich výskyt je ve střední Evropě relativně vzácný. Podobně jako v jiných nemalarických oblastech zastoupení ß-talasemie odráží historickou i současnou migraci a demografické změny, které ovlivňují genetickou variabilitu nynější populace žijící v této oblasti. Cíl: Stanovit frekvenci a spektrum mutací v ß-globinovém genu u pacientů z ČR a ze Slovenska s klinickými symptomy β- nebo β-talasemie. První část této studie byla publikována již téměř před dvaceti lety (Indrák et al., Hum Genet 1992; 88: ) a našim cílem bylo rozšířit a doplnit tuto původní práci. Pacienti a metody: Bylo analyzováno téměř 380 vzorků z hematologických center z České republiky a ze Slovenska. U všech vzorků byl vyšetřen krevní obraz, elektroforéza hemoglobinu (Hb) a byla provedena DNA analýza. Výsledky: Bylo identifikováno 22 ß-talasemických mutací u více jak 260 heterozygotů ze 152 nepříbuzných rodin českého nebo slovenského původu. Většina mutací (70 % pacientů) byla původem ze Středozemí. Nově objevená inzerce L1 retrotranspozonu do intronu ß-globinového genu představuje novou etiologii vzniku ß-talasemie. Vlivem inzerce došlo k umlčení exprese ß-globinového genu represivním chromatinem asociovaným s inzercí L1 (Piterkova et al, Heamatologica 2010; 95(s2): 417-8,(EHA abstrakt)). Na seznamu abnormálních hemoglobinů je v současnosti 14 ß-globinových variant zahrnujících i vzácnou variantu s vysokou afinitou ke kyslíku (Hb Olomouc) asociovanou s familiární polycytemií a dvě unikátní varianty vedoucí k hemolytické anemii s Heinzovými tělísky (nestabilní Hb Hradec Králové a Hb Haná). Závěr: V české a slovenské populaci je ß-talasemie vzácná, ale je nutné na ni pomýšlet v případech vrozené hypochromní mikrocytární anemie. Všichni pacienti s výjimkou čtyř byli heterozygoti s klinickými příznaky talasemie minor, jen ojediněle se jednalo o dominantně dědičnou formu ß-talasemie s fenotypovými projevy závažné talasemie minor nebo talasemie intermedia. Tři ze čtyř homozygotů nebo dvojitých heterozygotů ß-talasemie byli imigranti z malarických oblastí. Genetický posun a migrace v minulosti i v současnosti jsou zodpovědné za zanesení alel ze středomořských oblastí do naší populace s výjimkou některých mutací vzniklých místně u jednotlivých rodin. Podpořeno granty: IGA NT11208 a IGA NS /2009; MSM ; LF_2011_006 a LF_2011_ Úloha transkripčních faktorů Fli1, EKLF, PU.1, p53 a enzymu HDM2 u refrakterní anemie a megakaryopoezy u 5q- syndromu Neuwirtová Radana, Fuchs Ota, Jonášová Anna, Holická Monika, Vostrý Martin, Kostečka Arnošt, Hájková Hana, Čermák Jaroslav, Vondráková Jana, Hochová Ivana, Šišková Magdalena, Libiger Jiří, Šponerová Dana, Kadlčková Eva, Nováková Ludmila, Černá Olga, Čmejla Radek, Pospíšilová Dagmar, Seifertová Naďa, Březinová Jana, Michalová Kyra (I. interní klinika VFN 1. LF UK, Praha; ÚHKT, Praha; HOK FN, Olomouc; OKH FN Motol, Praha; OKH Masarykova nemocnice, Ústí nad Labem; HTO Baťova nemocnice, Zlín; Hem.klin. FN Královské Vinohrady, Praha; Dět- 24 Transfuze Hematol. dnes, 17, 2011

158
159 P /. He oglo i opatie v české a slove ské popula i Divoká M., Koláčková M., Lipert J., Novosadová A., Orviská M., Lapčíková A., Lá íková L., Parts hová M., Pospíšilová D., Divoký V., I drák K. (Hemato-o kologi ká kli ika, FN a LF UP, Olo ou ; Ústav iologie, LF UP, Olo ou ; Dětská kli ika, FN a LF UP, Olomouc) Hemoglobinopatie - talase ie tal a struktur í he oglo i ové H varia t - jsou ejrozšíře ější o oge í dědič é e o i a světě. Postihují % světové popula e. Výsk t tě hto t pů vroze ý h a e ií je ve střed í Evropě vzá ý, ale s i igra í popula e ze Středo oří a z vý hod í Asie jeji h počet roste. V posled í h lete h js e v české a slove ské popula i detekovali tal alel u ví e jak rodi a struktur í H varia t u té ěř rodi. Aktualizova é výsledk jsou pravidel ě preze tová a OHD. Dosud lo v ČR a SR ide tifiková o β-tal uta í u ví e jak heteroz gotů s projev tal i or, příp. tal i ter edia. Mezi i i lo ový h β-tal uta í. Nej ověji lo zjiště o, že i zer e retrotra spozo u do. i tro u β-glo i ového ge u je ový e ha iz e vz iku β-tal z utlu e í e prese z důvodu h per et la e DNA a dea et la e hro ati u v o lasti β-globin-l pro otoru Lá- íková et al, zaslá o k pu lika i. Zjistili jsme i 3 deleč í t p δ/β-tal. Vedle relativ ě častý h - Si ilské δ/β-tal a Hb Lepore jsme v roce 2012 ide tifikovali i vzá ou Makedo skou δ/β-tal dele i u pa ie ta původe ze Středo oří. Molekulár ě-ge eti ké a alýz pro ož ou příto ost α-tal uta í l dosud provede u e o ý h; u z i h la potvrze a deleč í for a α-tal, ezi i i i ový t p dele e regulač í oblasti HBA lokusu (HS- dele e. Kro ě osičů vzá ý h β-tal alel s do i a t í dědič ostí ají vši h i ostat í pa ie ti s β-tal heterozygoti pro mutovanou globinovou alelu fe ot p β-tal minor. Heteroz goti s α-tal dele í -α/αα,--/αα,-α/-α jsou kli i k as pto atičtí resp. ají fe ot p α-tal i or. Kli i k závaž é for tal i ter edia a ajor jsou v ČR a SR vzá é, přesto jsou v posled í h lete h zjišťová dík i igra i z alari ký h o lastí. )jistili js e jak ko i a e dvou t pů β-tal uta í u dvojitý h heteroz gotů, tak ko i a e α-tal- a α-tal-2 delece u pacienta s HbH a ko i a i H S s α-tal dele í u česko-afri ké rodi. V aší popula i js e ide tifikovali β- glo i ový h H varia t, ves ěs s esta il í H patií s projev he ol ti ké a é ie, ale i osiče vzá ý h varia t s v sokou afi itou H ke k slíku s fa iliár í pol té ií. V ro e js e poprvé v ČR diag ostikovali H C u i igra ta ze Středo oří. Před ěte ašeho sděle í je popis zají avý h případů vzá ý h vroze ý h a e ií s preze ta í kli i ký h a olekulár ě ge eti ký h výsledků včet ě popisu etodik. Podpoře o gra t : IGA NT, IGA NT, LF_ 13_004 a LF _2013_010.





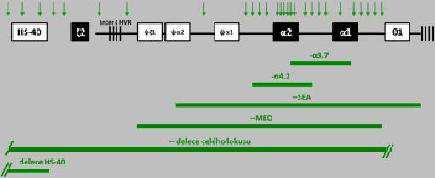


160
161 Příloha 6: Methemoglobinémie (domácí konference). Partschová, M., Mojzíková, R., Pospíšilová, P., Pospíšilová, D., Masárová, K., Divoký, V. Methemoglobinémie způsobená sníženou aktivitou cytochrom-b5-reduktázy: jedna nová mutace CYB5R3 genu. XXVI. Olomoucké hematologické dny, Olomouc, Sborník abstrakt, str. 71. Poster.

162
163 LabORaTORní hematologie 71 P37/ Methemoglobinémie způsobená sníženou aktivitou cytochrom-b5-reduktázy: jedna nová mutace CYb5R3 genu Partschová Martina, Mojzíková Renáta, Pospíšilová Pavla, Pospíšilová Dagmar, Masárová Katarína, Divoký Vladimír (Ústav biologie, LF UP; Dětská klinika, FN a LF UP, Olomouc CZE, Klinika hematológie a transfuziológie FNsP, Bratislava SVK) Úvod. Methemoglobin (MetHb) je za normálních podmínek v organismu odbouráván cytochromb5-reduktázou (Cb5R, NADH-methemoglobinreduktáza). Nedostatek Cb5R vede k vrozené autozomálně recesivní methemoglobinémii (RCM) projevující se zvýšenou hodnotou MetHb (> 1 %). Jsou známy 2 typy RCM: a) deicit Cb5R postihuje jen erytrocyty (Typ I); b) deicit postihuje všechny buňky (Typ II). Hlavním projevem RCM typu I je cyanóza (MetHb %), která je u RCM typu II doprovázená mentální retardací a neurologickými symptomy. Existuje ještě methemoglobinémie typu III, která je spojena s výskytem hemoglobinu (Hb) M a je autozomálně dominantní. Vrozené methemoglobinémie je nutné odlišit od těch získaných nebo asociovaných s přítomností nestabilního Hb. Pacienti a metody. 1. pacient: 15-letý chlapec s MetHb 7-11 %. Viditelným projevem bylo namodralé zbarvení kůže; účinky MetHb jsou redukovány kys. askorbovou. 2. pacient: náhodný záchyt 75-leté ženy s MetHb 11 % bez klinických projevů. V obou případech byla vyloučena hemoglobinopatie. Aktivita erytrocytární Cb5R byla stanovena spektrofotometricky pomocí artiiciálního substrátu ferrikyanidu. Genomická DNA byla použita pro sekvenační analýzu. Výsledky. U prvního pacienta byla stanovena snížená aktivita Cb5R (3 IU/g Hb; kontrolní rozmezí 8,26 12,39 IU/g Hb). Molekulárně-genetická analýza potvrdila, že je heterozygotem pro známý polymorizmus (c. G132A resp. p. P44P) a novou mutaci (A400G resp. p. M134V) v CYB5R3 genu. Asymptomatická matka má normální aktivitu Cb5R a není nositelkou ani jedné ze substitucí (otec není k dispozici). U druhé pacientky bylo prokázáno, že je heterozygotem pro substituci c. G890A tj. p. R297H. Stanovení aktivity Cb5R nebylo možné zajistit. Závěr. Mutace M134V se nachází na C-konci beta-řetězce FAD- -vazebné domény, který je spojen krátkou smyčkou s NADH-vazebnou doménou. Záměna by mohla vést k oslabení kontaktu mezi oběma doménami a ovlivnit tak stabilitu enzymu. Záměna R297H je situována ve FAD-vazebné doméně v blízkosti vazebného místa pro NADH a mohla by nepřímo ovlivňovat interakci mezi oběma kofaktory. Mutace M134V nebyla dosud popsána. Substituce c. G890A pro záměnu R297H je uváděna v databázi ( jako vzácný SNIP. Oba pacienti vykazují mírnou resp. asymptomatickou methemoglobinémii; v recesivní koniguraci bez dalších funkčních studií nelze kauzalitu mutací CYB5R3 genu jednoznačně hodnotit. Podpořeno MZ čr NT11208 a IGA UP LF_2012_016. P38/ Izolácia dna zo slín na hla typizáciu u leukopenických a transfúzne závislých pacientov Farkašová Denisa, Kušíková Mária, Stemnická Denisa, Al Sabty Firas, Skraková Marcela, Mistrík Martin (Klinika hematológie a transfuziológie, LFUK, SZU a UNB, Národný register darcov kostnej drene, Nemocnica sv. Cyrila a Metoda, Bratislava SVK) Úvod: u leukopenických a transfúzne závislých pacientov, ktorí sú indikovaní na alogénnu transplantáciu krvotvorných buniek (TKB) nie je vždy možné vyizolovať dostatočne koncentrovanú a čistú DNA zo vzorky krvi z dôvodu leukopénie a alokontaminácie leukocytmi darcov. HLA typizácia pacientov molekulovo biologickými metódami si vyžaduje dostatočné množstvo a koncentráciu DNA. Cieľ: zhodnotenie izolácie DNA zo slín u leukopenických a transfúzne závislých pacientov. Metódy: súbor našich pacientov tvorí 5 pacientov s hematoonkologickými ochoreniami, ktorí boli indikovaní na alogénnu TKB. Pre ťažkú leukopéniu nebolo možné vyizolovať DNA z periférnej krvi a preto sme zvolili alternatívnu metódu izolácie DNA zo slín použitím odberového kitu OG-510 od irmy DNA genotek. DNA sme izolovali podľa protokolu k danému kitu. Efektivita izolácie DNA bola hodnotená spektrofotometrickou analýzou a následne sa HLA typizácia vykonávala PCR-SSP metódou. Výsledky: použitím kitu OG-510 na izoláciu DNA z 2ml slín sme u všetkých pacientov vyizolovali 200µl DNA. Spektrofotometrická analýza ukázala, že koncentrácia DNA je od 122 ng/µl 214 ng/µl a index čistoty je od 1,56- do 1,7. Vyizolovaná DNA bola následne nariedená na koncentráciu 15 ng/µl

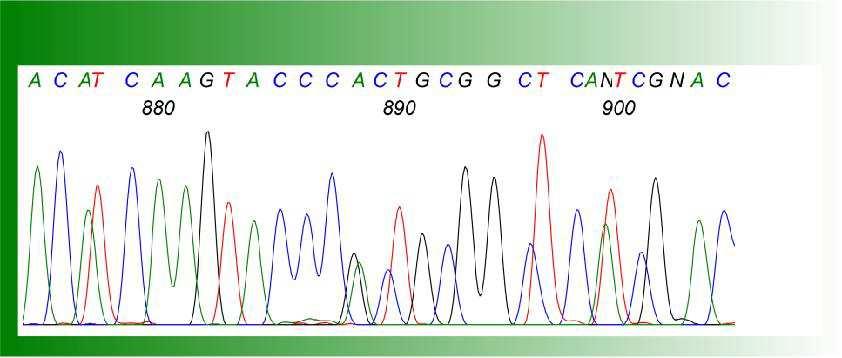
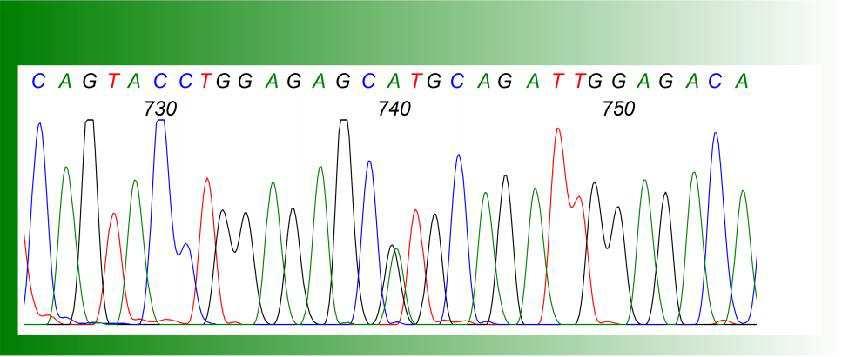


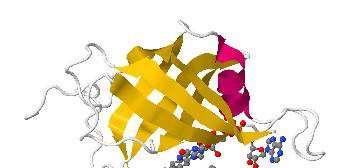

164
165 Příloha 7: Hemochromatóza (zahraniční konference). Pospisilova, P., Mojzikova, R., Partschova, M., Zidova, Z., Horvathova, M., Pospisilova, D., Divoky, V. Hemolytic anemia with hyperferritinemia in Czech pyruvate kinase deficient patient a novel homozygous frameshift deletion in PKLR gene (p. R518fs) combined with TFR2 (p.r752h) and SLC40A1 (IVS1-24 C>G) gene polymorphisms. 19th ERCS meeting. Forteiland, IJmuiden, The Netherlands, Abstract book, p.47. Přednáška.

166

167
MUDr. Kissová Jarmila, Ph.D. Oddělení klinické hematologie FN Brno
 MUDr. Kissová Jarmila, Ph.D. Oddělení klinické hematologie FN Brno Krvetvorba představuje proces tvorby krvinek v krvetvorných orgánech Krvetvorba je nesmírně komplikovaný, komplexně řízený a dodnes ne
MUDr. Kissová Jarmila, Ph.D. Oddělení klinické hematologie FN Brno Krvetvorba představuje proces tvorby krvinek v krvetvorných orgánech Krvetvorba je nesmírně komplikovaný, komplexně řízený a dodnes ne
Dr. Kissová Jarmila Oddělení klinické hematologie FN Brno
 Dr. Kissová Jarmila Oddělení klinické hematologie FN Brno Krvetvorba představuje proces tvorby krvinek v krvetvorných orgánech Krvetvorba je nesmírně komplikovaný, komplexně řízený a dodnes ne zcela dobře
Dr. Kissová Jarmila Oddělení klinické hematologie FN Brno Krvetvorba představuje proces tvorby krvinek v krvetvorných orgánech Krvetvorba je nesmírně komplikovaný, komplexně řízený a dodnes ne zcela dobře
Převzato na základě svolení Macmillan Publishers Ltd: Nat Rev Genet. 2001;2(4):245-55), copyright (2001).
:245-55), copyright (2001).") α-talasemie Autor: Alexandra Kredátusová Výskyt Talasemie jsou světově nejrozšířenější dědičně podmíněné krevní onemocnění. Nejčastější jsou v malarických oblastech (obr. 1), protože nositeli talasemického
α-talasemie Autor: Alexandra Kredátusová Výskyt Talasemie jsou světově nejrozšířenější dědičně podmíněné krevní onemocnění. Nejčastější jsou v malarických oblastech (obr. 1), protože nositeli talasemického
Vývoj krvetvorby. lení klinické hematologie FN Brno
 Vývoj krvetvorby Dr. Kissová Jarmila Oddělen lení klinické hematologie FN Brno Krvetvorba představuje p proces tvorby krvinek v krvetvorných orgánech. Krvetvorba je nesmírn rně komplikovaný, komplexně
Vývoj krvetvorby Dr. Kissová Jarmila Oddělen lení klinické hematologie FN Brno Krvetvorba představuje p proces tvorby krvinek v krvetvorných orgánech. Krvetvorba je nesmírn rně komplikovaný, komplexně
ANÉMIE Emanuel Nečas 2014
 ANÉMIE Emanuel Nečas 2014 necas@cesnet.cz Sylabus přednášky Anémie 1 1. Co je anémie 2. Co způsobuje anémii 3. Anemický syndrom 4. Klasifikace anémií 4. Shrnutí podstatných informací 1. Co je anémie? Anémie
ANÉMIE Emanuel Nečas 2014 necas@cesnet.cz Sylabus přednášky Anémie 1 1. Co je anémie 2. Co způsobuje anémii 3. Anemický syndrom 4. Klasifikace anémií 4. Shrnutí podstatných informací 1. Co je anémie? Anémie
Erytrocyty. Hemoglobin. Krevní skupiny a Rh faktor. Krevní transfúze. Somatologie Mgr. Naděžda Procházková
 Erytrocyty. Hemoglobin. Krevní skupiny a Rh faktor. Krevní transfúze. Somatologie Mgr. Naděžda Procházková Formované krevní elementy: Buněčné erytrocyty, leukocyty Nebuněčné trombocyty Tvorba krevních
Erytrocyty. Hemoglobin. Krevní skupiny a Rh faktor. Krevní transfúze. Somatologie Mgr. Naděžda Procházková Formované krevní elementy: Buněčné erytrocyty, leukocyty Nebuněčné trombocyty Tvorba krevních
Anémie. Bourková L., OKH FN Brno
 Anémie Bourková L., OKH FN Brno Vyšetření retikulocytů Barvení RNA v erytrocytech: mikroskopicky supravitální barvení (bez fixace preparátu) analyzátorem analýza prošlého světla analýza fluorescence Mikroskopické
Anémie Bourková L., OKH FN Brno Vyšetření retikulocytů Barvení RNA v erytrocytech: mikroskopicky supravitální barvení (bez fixace preparátu) analyzátorem analýza prošlého světla analýza fluorescence Mikroskopické
Exprese genetického kódu Centrální dogma molekulární biologie DNA RNA proteinu transkripce DNA mrna translace proteosyntéza
 Exprese genetického kódu Centrální dogma molekulární biologie - genetická informace v DNA -> RNA -> primárního řetězce proteinu 1) transkripce - přepis z DNA do mrna 2) translace - přeložení z kódu nukleových
Exprese genetického kódu Centrální dogma molekulární biologie - genetická informace v DNA -> RNA -> primárního řetězce proteinu 1) transkripce - přepis z DNA do mrna 2) translace - přeložení z kódu nukleových
Anémie. Bourková L., OKH FN Brno
 Anémie Bourková L., OKH FN Brno Sledování vyšetření Sledovat: hloubku anémie v KO morfologické změny erytrocytů v periferní krvi (barevné, tvarové, inkluze) morfologické a množstevní změny erytrocytární
Anémie Bourková L., OKH FN Brno Sledování vyšetření Sledovat: hloubku anémie v KO morfologické změny erytrocytů v periferní krvi (barevné, tvarové, inkluze) morfologické a množstevní změny erytrocytární
Exprese genetické informace
 Exprese genetické informace Tok genetické informace DNA RNA Protein (výjimečně RNA DNA) DNA RNA : transkripce RNA protein : translace Gen jednotka dědičnosti sekvence DNA nutná k produkci funkčního produktu
Exprese genetické informace Tok genetické informace DNA RNA Protein (výjimečně RNA DNA) DNA RNA : transkripce RNA protein : translace Gen jednotka dědičnosti sekvence DNA nutná k produkci funkčního produktu
ANÉMIE CHRONICKÝCH CHOROB
 ANÉMIE CHRONICKÝCH CHOROB (ACD anemia of chronic disease) seminář Martin Vokurka 2007 neoficiální verze pro studenty 2007 1 Proč se jí zabýváme? VELMI ČASTÁ!!! U hospitalizovaných pacientů je po sideropenii
ANÉMIE CHRONICKÝCH CHOROB (ACD anemia of chronic disease) seminář Martin Vokurka 2007 neoficiální verze pro studenty 2007 1 Proč se jí zabýváme? VELMI ČASTÁ!!! U hospitalizovaných pacientů je po sideropenii
AUG STOP AAAA S S. eukaryontní gen v genomové DNA. promotor exon 1 exon 2 exon 3 exon 4. kódující oblast. introny
 eukaryontní gen v genomové DNA promotor exon 1 exon 2 exon 3 exon 4 kódující oblast introny primární transkript (hnrna, pre-mrna) postranskripční úpravy (vznik maturované mrna) syntéza čepičky AUG vyštěpení
eukaryontní gen v genomové DNA promotor exon 1 exon 2 exon 3 exon 4 kódující oblast introny primární transkript (hnrna, pre-mrna) postranskripční úpravy (vznik maturované mrna) syntéza čepičky AUG vyštěpení
Co nám přináší migrace
 Projekt sponzorován z fondů FRVŠ 2334/2010 G3 Co nám přináší migrace Barbora Ludíková Dagmar Pospíšilová Dětská klinika Fakultní nemocnice Olomouc Lékařská fakulta Univerzita Palackého v Olomouci Hemolytické
Projekt sponzorován z fondů FRVŠ 2334/2010 G3 Co nám přináší migrace Barbora Ludíková Dagmar Pospíšilová Dětská klinika Fakultní nemocnice Olomouc Lékařská fakulta Univerzita Palackého v Olomouci Hemolytické
Základy molekulární biologie KBC/MBIOZ
 Základy molekulární biologie KBC/MBIOZ Mária Čudejková 2. Transkripce genu a její regulace Transkripce genetické informace z DNA na RNA Transkripce dvou genů zachycená na snímku z elektronového mikroskopu.
Základy molekulární biologie KBC/MBIOZ Mária Čudejková 2. Transkripce genu a její regulace Transkripce genetické informace z DNA na RNA Transkripce dvou genů zachycená na snímku z elektronového mikroskopu.
Beličková 1, J Veselá 1, E Stará 1, Z Zemanová 2, A Jonášová 2, J Čermák 1
 Beličková 1, J Veselá 1, E Stará 1, Z Zemanová 2, A Jonášová 2, J Čermák 1 1 Ústav hematologie a krevní transfuze, Praha 2 Všeobecná fakultní nemocnice, Praha MDS Myelodysplastický syndrom (MDS) je heterogenní
Beličková 1, J Veselá 1, E Stará 1, Z Zemanová 2, A Jonášová 2, J Čermák 1 1 Ústav hematologie a krevní transfuze, Praha 2 Všeobecná fakultní nemocnice, Praha MDS Myelodysplastický syndrom (MDS) je heterogenní
RIGORÓZNÍ OTÁZKY - BIOLOGIE ČLOVĚKA
 RIGORÓZNÍ OTÁZKY - BIOLOGIE ČLOVĚKA 1. Genotyp a jeho variabilita, mutace a rekombinace Specifická imunitní odpověď Prevence a časná diagnostika vrozených vad 2. Genotyp a prostředí Regulace buněčného
RIGORÓZNÍ OTÁZKY - BIOLOGIE ČLOVĚKA 1. Genotyp a jeho variabilita, mutace a rekombinace Specifická imunitní odpověď Prevence a časná diagnostika vrozených vad 2. Genotyp a prostředí Regulace buněčného
CZ.1.07/1.5.00/ Člověk a příroda
 GYMNÁZIUM TÝN NAD VLTAVOU, HAVLÍČKOVA 13 Číslo projektu Číslo a název šablony klíčové aktivity Tematická oblast CZ.1.07/1.5.00/34.0437 III/2- Inovace a zkvalitnění výuky prostřednictvím IVT Člověk a příroda
GYMNÁZIUM TÝN NAD VLTAVOU, HAVLÍČKOVA 13 Číslo projektu Číslo a název šablony klíčové aktivity Tematická oblast CZ.1.07/1.5.00/34.0437 III/2- Inovace a zkvalitnění výuky prostřednictvím IVT Člověk a příroda
VROZENÉ PORUCHY KRVETVORBY A JEJICH MANIFESTACE V DOSPĚLÉM VĚKU
 VROZENÉ PORUCHY KRVETVORBY A JEJICH MANIFESTACE V DOSPĚLÉM VĚKU Jaroslav Čermák Ústav hematologie a krevní transfuze, Praha VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY VROZENÉ
VROZENÉ PORUCHY KRVETVORBY A JEJICH MANIFESTACE V DOSPĚLÉM VĚKU Jaroslav Čermák Ústav hematologie a krevní transfuze, Praha VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY VROZENÉ
2. Z následujících tvrzení, týkajících se prokaryotické buňky, vyberte správné:
 Výběrové otázky: 1. Součástí všech prokaryotických buněk je: a) DNA, plazmidy b) plazmidy, mitochondrie c) plazmidy, ribozomy d) mitochondrie, endoplazmatické retikulum 2. Z následujících tvrzení, týkajících
Výběrové otázky: 1. Součástí všech prokaryotických buněk je: a) DNA, plazmidy b) plazmidy, mitochondrie c) plazmidy, ribozomy d) mitochondrie, endoplazmatické retikulum 2. Z následujících tvrzení, týkajících
Molekulární základy dědičnosti. Ústřední dogma molekulární biologie Struktura DNA a RNA
 Molekulární základy dědičnosti Ústřední dogma molekulární biologie Struktura DNA a RNA Ústřední dogma molekulární genetiky - vztah mezi nukleovými kyselinami a proteiny proteosyntéza replikace DNA RNA
Molekulární základy dědičnosti Ústřední dogma molekulární biologie Struktura DNA a RNA Ústřední dogma molekulární genetiky - vztah mezi nukleovými kyselinami a proteiny proteosyntéza replikace DNA RNA
DUM č. 10 v sadě. 37. Bi-2 Cytologie, molekulární biologie a genetika
 projekt GML Brno Docens DUM č. 10 v sadě 37. Bi-2 Cytologie, molekulární biologie a genetika Autor: Martin Krejčí Datum: 26.06.2014 Ročník: 6AF, 6BF Anotace DUMu: Procesy následující bezprostředně po transkripci.
projekt GML Brno Docens DUM č. 10 v sadě 37. Bi-2 Cytologie, molekulární biologie a genetika Autor: Martin Krejčí Datum: 26.06.2014 Ročník: 6AF, 6BF Anotace DUMu: Procesy následující bezprostředně po transkripci.
"Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Základy genetiky, základní pojmy
 "Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Základy genetiky, základní pojmy 1/75 Genetika = věda o dědičnosti Studuje biologickou informaci. Organizmy uchovávají,
"Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Základy genetiky, základní pojmy 1/75 Genetika = věda o dědičnosti Studuje biologickou informaci. Organizmy uchovávají,
Základní pojmy obecné genetiky, kvalitativní a kvantitativní znaky, vztahy mezi geny
 Obecná genetika Základní pojmy obecné genetiky, kvalitativní a kvantitativní znaky, vztahy mezi geny Doc. RNDr. Ing. Eva PALÁTOVÁ, PhD. Ing. Roman LONGAUER, CSc. Ústav zakládání a pěstění lesů LDF MENDELU
Obecná genetika Základní pojmy obecné genetiky, kvalitativní a kvantitativní znaky, vztahy mezi geny Doc. RNDr. Ing. Eva PALÁTOVÁ, PhD. Ing. Roman LONGAUER, CSc. Ústav zakládání a pěstění lesů LDF MENDELU
IMUNOGENETIKA I. Imunologie. nauka o obraných schopnostech organismu. imunitní systém heterogenní populace buněk lymfatické tkáně lymfatické orgány
 IMUNOGENETIKA I Imunologie nauka o obraných schopnostech organismu imunitní systém heterogenní populace buněk lymfatické tkáně lymfatické orgány lymfatická tkáň thymus Imunita reakce organismu proti cizorodým
IMUNOGENETIKA I Imunologie nauka o obraných schopnostech organismu imunitní systém heterogenní populace buněk lymfatické tkáně lymfatické orgány lymfatická tkáň thymus Imunita reakce organismu proti cizorodým
Myelodysplastický syndrom. MUDr.Kissová Jarmila Oddělení klinické hematologie FN Brno
 Myelodysplastický syndrom MUDr.Kissová Jarmila Oddělení klinické hematologie FN Brno Myelodysplázie Myelodysplázie- přítomnost morfologicky abnormální krvetvorby, má mnoho příčin ( deficit B 12, folátu,
Myelodysplastický syndrom MUDr.Kissová Jarmila Oddělení klinické hematologie FN Brno Myelodysplázie Myelodysplázie- přítomnost morfologicky abnormální krvetvorby, má mnoho příčin ( deficit B 12, folátu,
Genetická kontrola prenatáln. lního vývoje
 Genetická kontrola prenatáln lního vývoje Stádia prenatáln lního vývoje Preembryonální stádium do 6. dne po oplození zygota až blastocysta polární organizace cytoplasmatických struktur zygoty Embryonální
Genetická kontrola prenatáln lního vývoje Stádia prenatáln lního vývoje Preembryonální stádium do 6. dne po oplození zygota až blastocysta polární organizace cytoplasmatických struktur zygoty Embryonální
Genetický polymorfismus
 Genetický polymorfismus Za geneticky polymorfní je považován znak s nejméně dvěma geneticky podmíněnými variantami v jedné populaci, které se nachází v takových frekvencích, že i zřídkavá má frekvenci
Genetický polymorfismus Za geneticky polymorfní je považován znak s nejméně dvěma geneticky podmíněnými variantami v jedné populaci, které se nachází v takových frekvencích, že i zřídkavá má frekvenci
Základy genetiky 2a. Přípravný kurz Komb.forma studia oboru Všeobecná sestra
 Základy genetiky 2a Přípravný kurz Komb.forma studia oboru Všeobecná sestra Základní genetické pojmy: GEN - úsek DNA molekuly, který svojí primární strukturou určuje primární strukturu jiné makromolekuly
Základy genetiky 2a Přípravný kurz Komb.forma studia oboru Všeobecná sestra Základní genetické pojmy: GEN - úsek DNA molekuly, který svojí primární strukturou určuje primární strukturu jiné makromolekuly
Terapeutické klonování, náhrada tkání a orgánů
 Transfekce, elektroporace, retrovirová infekce Vnesení genů Vrstva fibroblastů, LIF Terapeutické klonování, náhrada tkání a orgánů Selekce ES buněk, v nichž došlo k začlenění vneseného genu homologní rekombinací
Transfekce, elektroporace, retrovirová infekce Vnesení genů Vrstva fibroblastů, LIF Terapeutické klonování, náhrada tkání a orgánů Selekce ES buněk, v nichž došlo k začlenění vneseného genu homologní rekombinací
HEMOPOESA. Periody krvetvorby, kmenové a progenitorové buňky; regulace hemopoesy. Ústav histologie a embryologie
 HEMOPOESA Periody krvetvorby, kmenové a progenitorové buňky; regulace hemopoesy Ústav histologie a embryologie MUDr. Radomíra Vagnerová, CSc. Předmět: Obecná histologie a obecná embryologie B02241 Přednášky
HEMOPOESA Periody krvetvorby, kmenové a progenitorové buňky; regulace hemopoesy Ústav histologie a embryologie MUDr. Radomíra Vagnerová, CSc. Předmět: Obecná histologie a obecná embryologie B02241 Přednášky
UNIVERZITA PALACKÉHO V OLOMOUCI. Lékařská fakulta. Ústav biologie
 UNIVERZITA PALACKÉHO V OLOMOUCI Lékařská fakulta Ústav biologie MOLEKULÁRNĚ-GENETICKÁ CHARAKTERIZACE TALASEMIÍ A HEMOGLOBINOVÝCH VARIANT V ČESKÉ A SLOVENSKÉ POPULACI Dizertační práce RNDr. Martina Divoká
UNIVERZITA PALACKÉHO V OLOMOUCI Lékařská fakulta Ústav biologie MOLEKULÁRNĚ-GENETICKÁ CHARAKTERIZACE TALASEMIÍ A HEMOGLOBINOVÝCH VARIANT V ČESKÉ A SLOVENSKÉ POPULACI Dizertační práce RNDr. Martina Divoká
Jaké máme leukémie? Akutní myeloidní leukémie (AML) Akutní lymfoblastická leukémie (ALL) Chronické leukémie, myelodysplastický syndrom,
 Akutní lymfoblastická leukémie (ALL) Chronické leukémie, myelodysplastický syndrom,") Akutní myeloidní leukémie (AML) Jaké máme leukémie? Akutní lymfoblastická leukémie (ALL) Chronické leukémie, myelodysplastický syndrom, Chronické leukémie, mnohočetný myelom, Někdy to není tak jednoznačné
Akutní myeloidní leukémie (AML) Jaké máme leukémie? Akutní lymfoblastická leukémie (ALL) Chronické leukémie, myelodysplastický syndrom, Chronické leukémie, mnohočetný myelom, Někdy to není tak jednoznačné
Zkušební testy z patologické fyziologie. Pavel Maruna a kolektiv. Recenzovali: prof. MUDr. Emanuel Nečas, DrSc. prof. MUDr. Jaroslav Veselý, CSc.
 Zkušební testy z patologické fyziologie Pavel Maruna a kolektiv Recenzovali: prof. MUDr. Emanuel Nečas, DrSc. prof. MUDr. Jaroslav Veselý, CSc. Autorský kolektiv: prof. MUDr. Pavel Maruna, CSc. doc. MUDr.
Zkušební testy z patologické fyziologie Pavel Maruna a kolektiv Recenzovali: prof. MUDr. Emanuel Nečas, DrSc. prof. MUDr. Jaroslav Veselý, CSc. Autorský kolektiv: prof. MUDr. Pavel Maruna, CSc. doc. MUDr.
Sylabus témat ke zkoušce z lékařské biologie a genetiky. Struktura, reprodukce a rekombinace virů (DNA viry, RNA viry), význam v medicíně
, význam v medicíně") Sylabus témat ke zkoušce z lékařské biologie a genetiky Buněčná podstata reprodukce a dědičnosti Struktura a funkce prokaryot Struktura, reprodukce a rekombinace virů (DNA viry, RNA viry), význam v medicíně
Sylabus témat ke zkoušce z lékařské biologie a genetiky Buněčná podstata reprodukce a dědičnosti Struktura a funkce prokaryot Struktura, reprodukce a rekombinace virů (DNA viry, RNA viry), význam v medicíně
TĚLNÍ TEKUTINY KREVNÍ ELEMENTY
 Mgr. Šárka Vopěnková Gymnázium, SOŠ a VOŠ Ledeč nad Sázavou VY_32_INOVACE_01_3_11_BI1 TĚLNÍ TEKUTINY KREVNÍ ELEMENTY KREVNÍ BUŇKY ČERVENÉ KRVINKY (ERYTROCYTY) Bikonkávní, bezjaderné buňky Zvýšený počet:
Mgr. Šárka Vopěnková Gymnázium, SOŠ a VOŠ Ledeč nad Sázavou VY_32_INOVACE_01_3_11_BI1 TĚLNÍ TEKUTINY KREVNÍ ELEMENTY KREVNÍ BUŇKY ČERVENÉ KRVINKY (ERYTROCYTY) Bikonkávní, bezjaderné buňky Zvýšený počet:
Chromosomy a karyotyp člověka
 Chromosomy a karyotyp člověka Chromosom - 1 a více - u eukaryotických buněk uložen v jádře karyotyp - soubor všech chromosomů v jádře jedné buňky - tvořen z vláknem chromatinem = DNA + histony - malé bazické
Chromosomy a karyotyp člověka Chromosom - 1 a více - u eukaryotických buněk uložen v jádře karyotyp - soubor všech chromosomů v jádře jedné buňky - tvořen z vláknem chromatinem = DNA + histony - malé bazické
Molekulárn. rní. biologie Struktura DNA a RNA
 Molekulárn rní základy dědičnosti Ústřední dogma molekulárn rní biologie Struktura DNA a RNA Ústřední dogma molekulárn rní genetiky - vztah mezi nukleovými kyselinami a proteiny proteosyntéza replikace
Molekulárn rní základy dědičnosti Ústřední dogma molekulárn rní biologie Struktura DNA a RNA Ústřední dogma molekulárn rní genetiky - vztah mezi nukleovými kyselinami a proteiny proteosyntéza replikace
Proteiny Genová exprese. 2013 Doc. MVDr. Eva Bártová, Ph.D.
 Proteiny Genová exprese 2013 Doc. MVDr. Eva Bártová, Ph.D. Bílkoviny (proteiny), 15% 1g = 17 kj Monomer = aminokyseliny aminová skupina karboxylová skupina α -uhlík postranní řetězec Znát obecný vzorec
Proteiny Genová exprese 2013 Doc. MVDr. Eva Bártová, Ph.D. Bílkoviny (proteiny), 15% 1g = 17 kj Monomer = aminokyseliny aminová skupina karboxylová skupina α -uhlík postranní řetězec Znát obecný vzorec
Nìkteré vzácnìjší formy hereditárních anémií vyskytující se v dospìlé populaci v ÈR a SR β talasemie a nestabilní hemoglobinové varianty
 128. internistický den XXI. Vanýskùv den, Brno 2005 Nìkteré vzácnìjší formy hereditárních anémií vyskytující se v dospìlé populaci v ÈR a SR β talasemie a nestabilní hemoglobinové varianty V. Divoký 1,2,
128. internistický den XXI. Vanýskùv den, Brno 2005 Nìkteré vzácnìjší formy hereditárních anémií vyskytující se v dospìlé populaci v ÈR a SR β talasemie a nestabilní hemoglobinové varianty V. Divoký 1,2,
Co nás učí nádory? Prof. RNDr. Jana Šmardová, CSc. Ústav patologie FN Brno Přírodovědecká a Lékařská fakulta MU Brno
 Co nás učí nádory? Prof. RNDr. Jana Šmardová, CSc. Ústav patologie FN Brno Přírodovědecká a Lékařská fakulta MU Brno Brno, 17.5.2011 Izidor (Easy Door) Osnova přednášky 1. Proč nás rakovina tolik zajímá?
Co nás učí nádory? Prof. RNDr. Jana Šmardová, CSc. Ústav patologie FN Brno Přírodovědecká a Lékařská fakulta MU Brno Brno, 17.5.2011 Izidor (Easy Door) Osnova přednášky 1. Proč nás rakovina tolik zajímá?
Crossing-over. over. synaptonemální komplex
 Genetické mapy Crossing-over over v průběhu profáze I meiózy princip rekombinace genetického materiálu mezi maternálním a paternálním chromosomem synaptonemální komplex zlomy a nová spojení chromatinových
Genetické mapy Crossing-over over v průběhu profáze I meiózy princip rekombinace genetického materiálu mezi maternálním a paternálním chromosomem synaptonemální komplex zlomy a nová spojení chromatinových
DMPK (ZNF9) V DIFERENCOVANÝCH. Z, Kroupová I, Falk M* M
 V DIFERENCOVANÝCH. Z, Kroupová I, Falk M* M") FISH ANALÝZA m-rna DMPK (ZNF9) V DIFERENCOVANÝCH TKÁNÍCH PACIENT IENTŮ S MYOTONICKOU DYSTROFI FIÍ Lukáš Z, Kroupová I, Falk M* M Ústav patologie FN Brno *Biofyzikáln lní ústav AVČR R Brno Definice MD Myotonická
FISH ANALÝZA m-rna DMPK (ZNF9) V DIFERENCOVANÝCH TKÁNÍCH PACIENT IENTŮ S MYOTONICKOU DYSTROFI FIÍ Lukáš Z, Kroupová I, Falk M* M Ústav patologie FN Brno *Biofyzikáln lní ústav AVČR R Brno Definice MD Myotonická
->Oba typy buněk mají paměť. V případě, že se v těle objeví např. stejný druh viru podruhé,
 1 KREV Krev je hlavní součástí vnitřního prostředí organismu. Je to tekutý orgán, který má dvě složky: složku tekutou (plazma) a buněčnou (leukocyty, erytrocyty, trombocyty). 1.1 FUNKCE KRVE Transportní
1 KREV Krev je hlavní součástí vnitřního prostředí organismu. Je to tekutý orgán, který má dvě složky: složku tekutou (plazma) a buněčnou (leukocyty, erytrocyty, trombocyty). 1.1 FUNKCE KRVE Transportní
BÍLKOVINY. V organismu se nedají nahradit jinými sloučeninami, jen jako zdroj energie je mohou nahradit sacharidy a lipidy.
 BÍLKOVINY o makromolekulární látky, z velkého počtu AMK zbytků o základ všech organismů o rostliny je vytvářejí z anorganických sloučenin (dusičnanů) o živočichové je musejí přijímat v potravě, v trávicím
BÍLKOVINY o makromolekulární látky, z velkého počtu AMK zbytků o základ všech organismů o rostliny je vytvářejí z anorganických sloučenin (dusičnanů) o živočichové je musejí přijímat v potravě, v trávicím
MYELOFIBROSA - DIAGNOSTIKA A LÉČEBNÉ MOŽNOSTI
 MYELOFIBROSA - DIAGNOSTIKA A LÉČEBNÉ MOŽNOSTI DEFINICE: chronické myeloproliferativní (klonální) onemocnění charakteristické zmnožením vaziva v kostní dřeni a extramedulární krvetvorbou (první popis -
MYELOFIBROSA - DIAGNOSTIKA A LÉČEBNÉ MOŽNOSTI DEFINICE: chronické myeloproliferativní (klonální) onemocnění charakteristické zmnožením vaziva v kostní dřeni a extramedulární krvetvorbou (první popis -
rní tekutinu (ECF), tj. cca 1/3 celkového množstv
, tj. cca 1/3 celkového množstv") Představují tzv. extracelulárn rní tekutinu (ECF), tj. cca 1/3 celkového množstv ství vody v tělet (voda tvoří 65-75% váhy v těla; t z toho 2/3 vody jsou vázanv zané intracelulárn rně) Lymfa (míza) Tkáňový
Představují tzv. extracelulárn rní tekutinu (ECF), tj. cca 1/3 celkového množstv ství vody v tělet (voda tvoří 65-75% váhy v těla; t z toho 2/3 vody jsou vázanv zané intracelulárn rně) Lymfa (míza) Tkáňový
Abnormality bílých krvinek. MUDr.Kissová Jarmila Oddělení klinické hematologie FN Brno
 Abnormality bílých krvinek MUDr.Kissová Jarmila Oddělení klinické hematologie FN Brno Abnormality bílých krvinek Kvantitativní poruchy leukocytů - reaktivní změny - choroby monocyto-makrofágového makrofágového
Abnormality bílých krvinek MUDr.Kissová Jarmila Oddělení klinické hematologie FN Brno Abnormality bílých krvinek Kvantitativní poruchy leukocytů - reaktivní změny - choroby monocyto-makrofágového makrofágového
von Willebrandova choroba Mgr. Jaroslava Machálková
 von Willebrandova choroba Mgr. Jaroslava Machálková von Willebrandova choroba -je dědičná krvácivá choroba způsobená vrozeným kvantitativním či kvalitativním defektem von Willebrandova faktoru postihuje
von Willebrandova choroba Mgr. Jaroslava Machálková von Willebrandova choroba -je dědičná krvácivá choroba způsobená vrozeným kvantitativním či kvalitativním defektem von Willebrandova faktoru postihuje
Hemofilie. Alena Štambachová, Jitka Šlechtová hematologický úsek ÚKBH FN v Plzni
 Hemofilie Alena Štambachová, Jitka Šlechtová hematologický úsek ÚKBH FN v Plzni Definice hemofilie Nevyléčitelná vrozená krvácivá choroba s nedostatkem plazmatických faktorů FVIII hemofile A FIX hemofile
Hemofilie Alena Štambachová, Jitka Šlechtová hematologický úsek ÚKBH FN v Plzni Definice hemofilie Nevyléčitelná vrozená krvácivá choroba s nedostatkem plazmatických faktorů FVIII hemofile A FIX hemofile
Buňky, tkáně, orgány, soustavy
 Lidská buňka buněčné organely a struktury: Jádro Endoplazmatické retikulum Goldiho aparát Mitochondrie Lysozomy Centrioly Cytoskelet Cytoplazma Cytoplazmatická membrána Buněčné jádro Jadérko Karyoplazma
Lidská buňka buněčné organely a struktury: Jádro Endoplazmatické retikulum Goldiho aparát Mitochondrie Lysozomy Centrioly Cytoskelet Cytoplazma Cytoplazmatická membrána Buněčné jádro Jadérko Karyoplazma
DUM č. 11 v sadě. 37. Bi-2 Cytologie, molekulární biologie a genetika
 projekt GML Brno Docens DUM č. 11 v sadě 37. Bi-2 Cytologie, molekulární biologie a genetika Autor: Martin Krejčí Datum: 30.06.2014 Ročník: 6AF, 6BF Anotace DUMu: Princip genové exprese, intenzita překladu
projekt GML Brno Docens DUM č. 11 v sadě 37. Bi-2 Cytologie, molekulární biologie a genetika Autor: Martin Krejčí Datum: 30.06.2014 Ročník: 6AF, 6BF Anotace DUMu: Princip genové exprese, intenzita překladu
Leukemie a myeloproliferativní onemocnění
 Leukemie a myeloproliferativní onemocnění Myeloproliferativní tumory Klonální onemocnění hematopoetických stem cells charakterizované proliferací jedné nebo více myeloidních řad. Dospělí peak 5. 7. dekáda,
Leukemie a myeloproliferativní onemocnění Myeloproliferativní tumory Klonální onemocnění hematopoetických stem cells charakterizované proliferací jedné nebo více myeloidních řad. Dospělí peak 5. 7. dekáda,
Exprese genetické informace
 Exprese genetické informace Stavební kameny nukleových kyselin Nukleotidy = báze + cukr + fosfát BÁZE FOSFÁT Nukleosid = báze + cukr CUKR Báze Cyklické sloučeniny obsahující dusík puriny nebo pyrimidiny
Exprese genetické informace Stavební kameny nukleových kyselin Nukleotidy = báze + cukr + fosfát BÁZE FOSFÁT Nukleosid = báze + cukr CUKR Báze Cyklické sloučeniny obsahující dusík puriny nebo pyrimidiny
V organismu se bílkoviny nedají nahradit žádnými jinými sloučeninami, jen jako zdroj energie je mohou nahradit sacharidy a lipidy.
 BÍLKOVINY Bílkoviny jsou biomakromolekulární látky, které se skládají z velkého počtu aminokyselinových zbytků. Vytvářejí látkový základ života všech organismů. V tkáních vyšších organismů a člověka je
BÍLKOVINY Bílkoviny jsou biomakromolekulární látky, které se skládají z velkého počtu aminokyselinových zbytků. Vytvářejí látkový základ života všech organismů. V tkáních vyšších organismů a člověka je
Krev, složení krve, formované krevní elementy
 Krev, složení krve, formované krevní elementy Ústav pro histologii a embryologii Předmět: Histologie a embryologie 1, B01131, obor Zubní lékařství Datum přednášky: 5.11.2013 SLOŽENÍ Celkový objem krve
Krev, složení krve, formované krevní elementy Ústav pro histologii a embryologii Předmět: Histologie a embryologie 1, B01131, obor Zubní lékařství Datum přednášky: 5.11.2013 SLOŽENÍ Celkový objem krve
Akutní leukémie a myelodysplastický syndrom. Hemato-onkologická klinika FN a LF UP Olomouc
 Akutní leukémie a myelodysplastický syndrom Hemato-onkologická klinika FN a LF UP Olomouc Akutní leukémie (AL) Představují heterogenní skupinu chorob charakterizovaných kumulací klonu nevyzrálých, nádorově
Akutní leukémie a myelodysplastický syndrom Hemato-onkologická klinika FN a LF UP Olomouc Akutní leukémie (AL) Představují heterogenní skupinu chorob charakterizovaných kumulací klonu nevyzrálých, nádorově
Genová etiologie nemocí
 Genová etiologie nemocí 1. Obecná etiologie nemocí 1. Obecná etiologie nemocí 2. Mutace genů v germinativních a somatických buňkách 3. Molekulární fyziologie genu 4. Regulace aktivity genu (genové exprese)
Genová etiologie nemocí 1. Obecná etiologie nemocí 1. Obecná etiologie nemocí 2. Mutace genů v germinativních a somatických buňkách 3. Molekulární fyziologie genu 4. Regulace aktivity genu (genové exprese)
Nukleové kyseliny. Nukleové kyseliny. Genetická informace. Gen a genom. Složení nukleových kyselin. Centrální dogma molekulární biologie
 Centrální dogma molekulární biologie ukleové kyseliny 1865 zákony dědičnosti (Johann Gregor Transkripce D R Translace rotein Mendel) Replikace 1869 objev nukleových kyselin (Miescher) 1944 nukleové kyseliny
Centrální dogma molekulární biologie ukleové kyseliny 1865 zákony dědičnosti (Johann Gregor Transkripce D R Translace rotein Mendel) Replikace 1869 objev nukleových kyselin (Miescher) 1944 nukleové kyseliny
Krvetvorba. doc. MUDr. Julie Bienertová Vašků, Ph.D. Ústav patologické fyziologie LF MU CZ.1.07/2.2.00/
 doc. MUDr. Julie Bienertová Vašků, Ph.D. Ústav patologické fyziologie LF MU Krvetvorba CZ.1.07/2.2.00/28.0041 Centrum interaktivních a multimediálních studijních opor pro inovaci výuky a efektivní učení
doc. MUDr. Julie Bienertová Vašků, Ph.D. Ústav patologické fyziologie LF MU Krvetvorba CZ.1.07/2.2.00/28.0041 Centrum interaktivních a multimediálních studijních opor pro inovaci výuky a efektivní učení
b) Jak se změní sekvence aminokyselin v polypeptidu, pokud dojde v pozici 23 k záměně bázového páru GC za TA (bodová mutace) a s jakými následky?
 Jak se změní sekvence aminokyselin v polypeptidu, pokud dojde v pozici 23 k záměně bázového páru GC za TA (bodová mutace) a s jakými následky?") 1.1: Gén pro polypeptid, který je součástí peroxidázy buku lesního, má sekvenci 3'...TTTACAGTCCATTCGACTTAGGGGCTAAGGTACCTGGAGCCCACGTTTGGGTCATCCAG...5' 5'...AAATGTCAGGTAAGCTGAATCCCCGATTCCATGGACCTCGGGTGCAAACCCAGTAGGTC...3'
1.1: Gén pro polypeptid, který je součástí peroxidázy buku lesního, má sekvenci 3'...TTTACAGTCCATTCGACTTAGGGGCTAAGGTACCTGGAGCCCACGTTTGGGTCATCCAG...5' 5'...AAATGTCAGGTAAGCTGAATCCCCGATTCCATGGACCTCGGGTGCAAACCCAGTAGGTC...3'
Výskyt MHC molekul. RNDr. Ivana Fellnerová, Ph.D. ajor istocompatibility omplex. Funkce MHC glykoproteinů
 RNDr. Ivana Fellnerová, Ph.D. Katedra zoologie, PřF UP Olomouc = ajor istocompatibility omplex Skupina genů na 6. chromozomu (u člověka) Kódují membránové glykoproteiny, tzv. MHC molekuly, MHC molekuly
RNDr. Ivana Fellnerová, Ph.D. Katedra zoologie, PřF UP Olomouc = ajor istocompatibility omplex Skupina genů na 6. chromozomu (u člověka) Kódují membránové glykoproteiny, tzv. MHC molekuly, MHC molekuly
Tematické okruhy k SZZ v bakalářském studijním oboru Zdravotní laborant bakalářského studijního programu B5345 Specializace ve zdravotnictví
 Tematické okruhy k SZZ v bakalářském studijním oboru Zdravotní laborant bakalářského studijního programu B5345 Specializace ve zdravotnictví Dle čl. 7 odst. 2 Směrnice děkana pro realizaci bakalářských
Tematické okruhy k SZZ v bakalářském studijním oboru Zdravotní laborant bakalářského studijního programu B5345 Specializace ve zdravotnictví Dle čl. 7 odst. 2 Směrnice děkana pro realizaci bakalářských
Mgr. Veronika Peňásová vpenasova@fnbrno.cz Laboratoř molekulární diagnostiky, OLG FN Brno Klinika dětské onkologie, FN Brno
 Retinoblastom Mgr. Veronika Peňásová vpenasova@fnbrno.cz Laboratoř molekulární diagnostiky, OLG FN Brno Klinika dětské onkologie, FN Brno Retinoblastom (RBL) zhoubný nádor oka, pocházející z primitivních
Retinoblastom Mgr. Veronika Peňásová vpenasova@fnbrno.cz Laboratoř molekulární diagnostiky, OLG FN Brno Klinika dětské onkologie, FN Brno Retinoblastom (RBL) zhoubný nádor oka, pocházející z primitivních
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/
 Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky Populační genetika (KBB/PG)
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky Populační genetika (KBB/PG)
Centrální dogma molekulární biologie
 řípravný kurz LF MU 2011/12 Centrální dogma molekulární biologie Nukleové kyseliny 1865 zákony dědičnosti (Johann Gregor Mendel) 1869 objev nukleových kyselin (Miescher) 1944 genetická informace v nukleových
řípravný kurz LF MU 2011/12 Centrální dogma molekulární biologie Nukleové kyseliny 1865 zákony dědičnosti (Johann Gregor Mendel) 1869 objev nukleových kyselin (Miescher) 1944 genetická informace v nukleových
Degenerace genetického kódu
 AJ: degeneracy x degeneration CJ: degenerace x degenerace Degenerace genetického kódu Genetický kód je degenerovaný, resp. redundantní, což znamená, že dva či více kodonů může kódovat jednu a tutéž aminokyselinu.
AJ: degeneracy x degeneration CJ: degenerace x degenerace Degenerace genetického kódu Genetický kód je degenerovaný, resp. redundantní, což znamená, že dva či více kodonů může kódovat jednu a tutéž aminokyselinu.
Bílkoviny a rostlinná buňka
 Bílkoviny a rostlinná buňka Bílkoviny Rostliny --- kontinuální diferenciace vytváření orgánů: - mitotická dělení -zvětšování buněk a tvorba buněčné stěny syntéza bílkovin --- fotosyntéza syntéza bílkovin
Bílkoviny a rostlinná buňka Bílkoviny Rostliny --- kontinuální diferenciace vytváření orgánů: - mitotická dělení -zvětšování buněk a tvorba buněčné stěny syntéza bílkovin --- fotosyntéza syntéza bílkovin
7. Regulace genové exprese, diferenciace buněk a epigenetika
 7. Regulace genové exprese, diferenciace buněk a epigenetika Aby mohl mnohobuněčný organismus efektivně fungovat, je třeba, aby se jednotlivé buňky specializovaly na určité funkce. Nový jedinec přitom
7. Regulace genové exprese, diferenciace buněk a epigenetika Aby mohl mnohobuněčný organismus efektivně fungovat, je třeba, aby se jednotlivé buňky specializovaly na určité funkce. Nový jedinec přitom
Glosář - Cestina. Odchylka počtu chromozomů v jádře buňky od normy. Např. 45 nebo 47 chromozomů místo obvyklých 46. Příkladem je trizomie 21
 Glosář - Cestina alely aneuploidie asistovaná reprodukce autozomálně dominantní autozomálně recesivní BRCA chromozom chromozomová aberace cytogenetický laborant de novo Různé formy genu, které se nacházejí
Glosář - Cestina alely aneuploidie asistovaná reprodukce autozomálně dominantní autozomálně recesivní BRCA chromozom chromozomová aberace cytogenetický laborant de novo Různé formy genu, které se nacházejí
OBRANNÝ IMUNITNÍ SYSTÉM
 Mgr. Šárka Vopěnková Gymnázium, SOŠ a VOŠ Ledeč nad Sázavou VY_32_INOVACE_02_3_04_BI2 OBRANNÝ IMUNITNÍ SYSTÉM Základní znaky: není vrozená specificky rozpoznává cizorodé látky ( antigeny) vyznačuje se
Mgr. Šárka Vopěnková Gymnázium, SOŠ a VOŠ Ledeč nad Sázavou VY_32_INOVACE_02_3_04_BI2 OBRANNÝ IMUNITNÍ SYSTÉM Základní znaky: není vrozená specificky rozpoznává cizorodé látky ( antigeny) vyznačuje se
Vytvořilo Oddělení lékařské genetiky FN Brno
 GONOSOMY GONOSOMY CHROMOSOMY X, Y Obr. 1 (Nussbaum, 2004) autosomy v chromosomovém páru homologní po celé délce chromosomů crossingover MEIÓZA Obr. 2 (Nussbaum, 2004) GONOSOMY CHROMOSOMY X, Y ODLIŠNOSTI
GONOSOMY GONOSOMY CHROMOSOMY X, Y Obr. 1 (Nussbaum, 2004) autosomy v chromosomovém páru homologní po celé délce chromosomů crossingover MEIÓZA Obr. 2 (Nussbaum, 2004) GONOSOMY CHROMOSOMY X, Y ODLIŠNOSTI
Hematologické laboratorní metody. Krevní obraz Koagulace Imunohematologie Podání krevní transfuze
 Hematologické laboratorní metody Krevní obraz Koagulace Imunohematologie Podání krevní transfuze Krevní obraz I leukocyty WBC (white blood cells) norma 4,0 9,0x 10 9 /l leukopenie útlum polékový, toxický,
Hematologické laboratorní metody Krevní obraz Koagulace Imunohematologie Podání krevní transfuze Krevní obraz I leukocyty WBC (white blood cells) norma 4,0 9,0x 10 9 /l leukopenie útlum polékový, toxický,
Diagnostika genetických změn u papilárního karcinomu štítné žlázy
 Diagnostika genetických změn u papilárního karcinomu štítné žlázy Vlasta Sýkorová Oddělení molekulární endokrinologie Endokrinologický ústav, Praha Nádory štítné žlázy folikulární buňka parafolikulární
Diagnostika genetických změn u papilárního karcinomu štítné žlázy Vlasta Sýkorová Oddělení molekulární endokrinologie Endokrinologický ústav, Praha Nádory štítné žlázy folikulární buňka parafolikulární
Anémie u chronických onemocnění
 Projekt sponzorován z fondů FRVŠ 2334/2010 G3 Anémie u chronických onemocnění Dagmar Pospíšilová Barbora Ludíková Dětská klinika Fakultní nemocnice Olomouc Lékařská fakulta Univerzita Palackého v Olomouci
Projekt sponzorován z fondů FRVŠ 2334/2010 G3 Anémie u chronických onemocnění Dagmar Pospíšilová Barbora Ludíková Dětská klinika Fakultní nemocnice Olomouc Lékařská fakulta Univerzita Palackého v Olomouci
1. Téma : Genetika shrnutí Název DUMu : VY_32_INOVACE_29_SPSOA_BIO_1_CHAM 2. Vypracovala : Hana Chamulová 3. Vytvořeno v projektu EU peníze středním
 1. Téma : Genetika shrnutí Název DUMu : VY_32_INOVACE_29_SPSOA_BIO_1_CHAM 2. Vypracovala : Hana Chamulová 3. Vytvořeno v projektu EU peníze středním školám Genetika - shrnutí TL2 1. Doplň: heterozygot,
1. Téma : Genetika shrnutí Název DUMu : VY_32_INOVACE_29_SPSOA_BIO_1_CHAM 2. Vypracovala : Hana Chamulová 3. Vytvořeno v projektu EU peníze středním školám Genetika - shrnutí TL2 1. Doplň: heterozygot,
Garant předmětu GEN: prof. Ing. Jindřich Čítek, CSc. Garant předmětu GEN1: prof. Ing. Václav Řehout, CSc.
 Garant předmětu GEN: prof. Ing. Jindřich Čítek, CSc. Garant předmětu GEN1: prof. Ing. Václav Řehout, CSc. Další vyučující: Ing. l. Večerek, PhD., Ing. L. Hanusová, Ph.D., Ing. L. Tothová Předpoklady: znalosti
Garant předmětu GEN: prof. Ing. Jindřich Čítek, CSc. Garant předmětu GEN1: prof. Ing. Václav Řehout, CSc. Další vyučující: Ing. l. Večerek, PhD., Ing. L. Hanusová, Ph.D., Ing. L. Tothová Předpoklady: znalosti
Těsně před infarktem. Jak předpovědět infarkt pomocí informatických metod. Jan Kalina, Marie Tomečková
 Těsně před infarktem Jak předpovědět infarkt pomocí informatických metod Jan Kalina, Marie Tomečková Program, osnova sdělení 13,30 Úvod 13,35 Stručně o ateroskleróze 14,15 Měření genových expresí 14,00
Těsně před infarktem Jak předpovědět infarkt pomocí informatických metod Jan Kalina, Marie Tomečková Program, osnova sdělení 13,30 Úvod 13,35 Stručně o ateroskleróze 14,15 Měření genových expresí 14,00
Hierarchy of hematopoietic differentiation.
 Hierarchy of hematopoietic differentiation. Vývoj erytrocytu ANÉMIE - definice, rozdělení DEFINICE: Snížení množství Hb ( zpravidla i Htk a počtu ery ) v 1 litru krve pod dolní hodnotu zdravých jedinců.
Hierarchy of hematopoietic differentiation. Vývoj erytrocytu ANÉMIE - definice, rozdělení DEFINICE: Snížení množství Hb ( zpravidla i Htk a počtu ery ) v 1 litru krve pod dolní hodnotu zdravých jedinců.
(II.) Určení krevní skupiny sklíčkovou metodou
 Určení krevní skupiny sklíčkovou metodou") (I.) Stanovení červeného krevního obrazu (II.) Určení krevní skupiny sklíčkovou metodou Fyziologie I - cvičení Fyziologický ústav LF MU, 2015 Michal Hendrych Červená krvinka erytrocyt (ery) bezjaderná
(I.) Stanovení červeného krevního obrazu (II.) Určení krevní skupiny sklíčkovou metodou Fyziologie I - cvičení Fyziologický ústav LF MU, 2015 Michal Hendrych Červená krvinka erytrocyt (ery) bezjaderná
Morbus von Willebrand
 Morbus von Willebrand 08:48 Bohumír Blažek Morbus von Willebrand nejčastější krvácivá choroba postihuje asi 1 % světové populace mírné symptomy, část nediagnostikována některé formy s těžkým průběhem zvláště
Morbus von Willebrand 08:48 Bohumír Blažek Morbus von Willebrand nejčastější krvácivá choroba postihuje asi 1 % světové populace mírné symptomy, část nediagnostikována některé formy s těžkým průběhem zvláště
Krev přednáška 1 fyzioterapie
 Krev přednáška 1 fyzioterapie Mgr. Helena Smítková Krev I 1 Krev Suspenze formovaných krevních elementů v plasmě (RBC, WBC, TRO) Dospělý 4,5-6 litrů (7-10% hmotnosti) Transport: O2, CO2, živiny glc, AK,
Krev přednáška 1 fyzioterapie Mgr. Helena Smítková Krev I 1 Krev Suspenze formovaných krevních elementů v plasmě (RBC, WBC, TRO) Dospělý 4,5-6 litrů (7-10% hmotnosti) Transport: O2, CO2, živiny glc, AK,
VÝZNAM FYZIOLOGICKÉ OBNOVY BUNĚK V MEDICÍNĚ
 OBNOVA A REPARACE 1 VÝZNAM FYZIOLOGICKÉ OBNOVY BUNĚK V MEDICÍNĚ Příklad: Fyziologická obnova buněk: obnova erytrocytů Rychlost obnovy: 2 miliony nových erytrocytů/s (při průměrné době života erytrocytu
OBNOVA A REPARACE 1 VÝZNAM FYZIOLOGICKÉ OBNOVY BUNĚK V MEDICÍNĚ Příklad: Fyziologická obnova buněk: obnova erytrocytů Rychlost obnovy: 2 miliony nových erytrocytů/s (při průměrné době života erytrocytu
ÚVOD DO TRANSPLANTAČNÍ IMUNOLOGIE
 ÚVOD DO TRANSPLANTAČNÍ IMUNOLOGIE Základní funkce imunitního systému Chrání integritu organizmu proti škodlivinám zevního a vnitřního původu: chrání organizmus proti patogenním mikroorganizmům a jejich
ÚVOD DO TRANSPLANTAČNÍ IMUNOLOGIE Základní funkce imunitního systému Chrání integritu organizmu proti škodlivinám zevního a vnitřního původu: chrání organizmus proti patogenním mikroorganizmům a jejich
Játra a imunitní systém
 Ústav klinické imunologie a alergologie LF MU, RECETOX, PřF Masarykovy univerzity, FN u sv. Anny v Brně, Pekařská 53, 656 91 Brno Játra a imunitní systém Vojtěch Thon vojtech.thon@fnusa.cz Výběr 5. Fórum
Ústav klinické imunologie a alergologie LF MU, RECETOX, PřF Masarykovy univerzity, FN u sv. Anny v Brně, Pekařská 53, 656 91 Brno Játra a imunitní systém Vojtěch Thon vojtech.thon@fnusa.cz Výběr 5. Fórum
Experimentáln navozený radia ní syndrom u pokusného zví ete 2
 Experimentáln navozený radia ní syndrom u pokusného zví ete 2 Hemopoéza Hierarchie kmenových bun k Erytropoéza http://www.noblesmedart.com/morph.mov Vyzrávání erytrocyt erytropoetin, Fe, kys. listová,
Experimentáln navozený radia ní syndrom u pokusného zví ete 2 Hemopoéza Hierarchie kmenových bun k Erytropoéza http://www.noblesmedart.com/morph.mov Vyzrávání erytrocyt erytropoetin, Fe, kys. listová,
A. chromozómy jsou rozděleny na 2 chromatidy spojené jen v místě centromery. B. vlákna dělícího vřeténka jsou připojena k chromozómům
 Karlova univerzita, Lékařská fakulta Hradec Králové Obor: všeobecné lékařství - test z biologie Vyberte tu z nabídnutých odpovědí (1-5), která je nejúplnější. Otázka Odpověď 1. Mezi organely membránového
Karlova univerzita, Lékařská fakulta Hradec Králové Obor: všeobecné lékařství - test z biologie Vyberte tu z nabídnutých odpovědí (1-5), která je nejúplnější. Otázka Odpověď 1. Mezi organely membránového
rodokmeny vazby mezi členy rodiny + popis pro konkrétní sledovaný znak využití Mendelových zákonů v lékařství genetické konzultace o možném výskytu
 Genealogie Monogenní dědičnost rodokmeny vazby mezi členy rodiny + popis pro konkrétní sledovaný znak využití Mendelových zákonů v lékařství genetické konzultace o možném výskytu onemocnění v rodině Genealogické
Genealogie Monogenní dědičnost rodokmeny vazby mezi členy rodiny + popis pro konkrétní sledovaný znak využití Mendelových zákonů v lékařství genetické konzultace o možném výskytu onemocnění v rodině Genealogické
EPIGENETIKA reverzibilních změn funkce genů, Epigenetické faktory ovlivňují fenotyp bez změny genotypu. Epigenetická
 EPIGENETIKA Epigenetika se zabývá studiem reverzibilních změn funkce genů, aniž by při tom došlo ke změnám v sekvenci jaderné DNA. Epigenetické faktory ovlivňují fenotyp bez změny genotypu. Epigenetická
EPIGENETIKA Epigenetika se zabývá studiem reverzibilních změn funkce genů, aniž by při tom došlo ke změnám v sekvenci jaderné DNA. Epigenetické faktory ovlivňují fenotyp bez změny genotypu. Epigenetická
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/
 Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky Populační genetika (KBB/PG)
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky Populační genetika (KBB/PG)
ve srovnání s eukaryoty (životnost v řádu hodin) u prokaryot kratší (životnost v řádu minut) na životnost / stabilitu molekuly mají vliv
 u prokaryot kratší (životnost v řádu minut) na životnost / stabilitu molekuly mají vliv") Urbanová Anna ve srovnání s eukaryoty (životnost v řádu hodin) u prokaryot kratší (životnost v řádu minut) na životnost / stabilitu molekuly mají vliv strukturní rysy mrna proces degradace každá mrna v
Urbanová Anna ve srovnání s eukaryoty (životnost v řádu hodin) u prokaryot kratší (životnost v řádu minut) na životnost / stabilitu molekuly mají vliv strukturní rysy mrna proces degradace každá mrna v
Biologie buňky. systém schopný udržovat se a rozmnožovat
 Biologie buňky 1665 - Robert Hook (korek, cellulae = buňka) Cytologie - věda zabývající se studiem buňek Buňka ozákladní funkční a stavební jednotka živých organismů onejmenší známý uspořádaný dynamický
Biologie buňky 1665 - Robert Hook (korek, cellulae = buňka) Cytologie - věda zabývající se studiem buňek Buňka ozákladní funkční a stavební jednotka živých organismů onejmenší známý uspořádaný dynamický
Protinádorová imunita. Jiří Jelínek
 Protinádorová imunita Jiří Jelínek Imunitní systém vs. nádor l imunitní systém je poslední přirozený nástroj organismu jak eliminovat vlastní buňky které se vymkly kontrole l do boje proti nádorovým buňkám
Protinádorová imunita Jiří Jelínek Imunitní systém vs. nádor l imunitní systém je poslední přirozený nástroj organismu jak eliminovat vlastní buňky které se vymkly kontrole l do boje proti nádorovým buňkám
Onemocnění krve. Krvetvorba, základní charakteristiky krve
 Onemocnění krve Krvetvorba, základní charakteristiky krve Krev Vysoce specializovaná tělesná tekutina Je důležitým spojovacím a transportním systémem Zajišťuje nepřetržitou výměnu látek mezi buňkami Napomáhá
Onemocnění krve Krvetvorba, základní charakteristiky krve Krev Vysoce specializovaná tělesná tekutina Je důležitým spojovacím a transportním systémem Zajišťuje nepřetržitou výměnu látek mezi buňkami Napomáhá
Izolace RNA. doc. RNDr. Jan Vondráček, PhD..
 Izolace RNA doc. RNDr. Jan Vondráček, PhD.. Metodiky izolace RNA celková buněčná RNA ( total RNA) zahrnuje řadu typů RNA, které se mohou lišit svými fyzikálněchemickými vlastnostmi a tedy i nároky na jejich
Izolace RNA doc. RNDr. Jan Vondráček, PhD.. Metodiky izolace RNA celková buněčná RNA ( total RNA) zahrnuje řadu typů RNA, které se mohou lišit svými fyzikálněchemickými vlastnostmi a tedy i nároky na jejich
Progrese HIV infekce z pohledu laboratorní imunologie
 Progrese HIV infekce z pohledu laboratorní imunologie 1 Lochmanová A., 2 Olbrechtová L., 2 Kolčáková J., 2 Zjevíková A. 1 OIA ZÚ Ostrava 2 klinika infekčních nemocí, FN Ostrava HIV infekce onemocnění s
Progrese HIV infekce z pohledu laboratorní imunologie 1 Lochmanová A., 2 Olbrechtová L., 2 Kolčáková J., 2 Zjevíková A. 1 OIA ZÚ Ostrava 2 klinika infekčních nemocí, FN Ostrava HIV infekce onemocnění s
NÁLEZ DVOJITĚ POZITIVNÍCH T LYMFOCYTŮ - CO TO MŮŽE ZNAMENAT? Ondřej Souček Ústav klinické imunologie a alergologie Fakultní nemocnice Hradec Králové
 NÁLEZ DVOJITĚ POZITIVNÍCH T LYMFOCYTŮ - CO TO MŮŽE ZNAMENAT? Ondřej Souček Ústav klinické imunologie a alergologie Fakultní nemocnice Hradec Králové LEUKÉMIE x LYMFOM Nádorová onemocnění buněk krvetvorné
NÁLEZ DVOJITĚ POZITIVNÍCH T LYMFOCYTŮ - CO TO MŮŽE ZNAMENAT? Ondřej Souček Ústav klinické imunologie a alergologie Fakultní nemocnice Hradec Králové LEUKÉMIE x LYMFOM Nádorová onemocnění buněk krvetvorné
19.b - Metabolismus nukleových kyselin a proteosyntéza
 19.b - Metabolismus nukleových kyselin a proteosyntéza Proteosyntéza vyžaduje především zajištění primární struktury. Informace je uložena v DNA (ev. RNA u některých virů) trvalá forma. Forma uskladnění
19.b - Metabolismus nukleových kyselin a proteosyntéza Proteosyntéza vyžaduje především zajištění primární struktury. Informace je uložena v DNA (ev. RNA u některých virů) trvalá forma. Forma uskladnění
Genetické aspekty vrozených vad metabolismu
 Genetické aspekty vrozených vad metabolismu Doc. MUDr. Alena Šantavá, CSc. Ústav lékařské genetiky a fetální medicíny FN a LF UP Olomouc Johann Gregor Mendel (1822-1884) Sir Archibald Garrod britský pediatr
Genetické aspekty vrozených vad metabolismu Doc. MUDr. Alena Šantavá, CSc. Ústav lékařské genetiky a fetální medicíny FN a LF UP Olomouc Johann Gregor Mendel (1822-1884) Sir Archibald Garrod britský pediatr
Klinické a molekulární aspekty poruch metabolismu železa seminář Martin Vokurka
 Klinické a molekulární aspekty poruch metabolismu železa seminář Martin Vokurka Železo je 4. nejhojnější prvek kůry zemské Využívají ho všechny živé organismy Co od něho chtějí? Jaké má tedy železo funkce?
Klinické a molekulární aspekty poruch metabolismu železa seminář Martin Vokurka Železo je 4. nejhojnější prvek kůry zemské Využívají ho všechny živé organismy Co od něho chtějí? Jaké má tedy železo funkce?