Midlle.gif (12243 bytes)
|
|
|
- Viktor Konečný
- před 8 lety
- Počet zobrazení:
Transkript
1 Left.gif (10785 Midlle.gif (12243 Right.gif (10788 Lidské prionové nemoci První zkušenosti a přehled literatury Human Prion Diseases František Koukolík, Radoslav Matěj Národní referenční laboratoř TSE/CJN Oddělení patologie Fakultní Thomayerovy nemocnice s poliklinikou, Praha Souhrn Prionové choroby jsou skupina neurodegenerativních onemocnění postihujících zvířata i lidi. Lidské prionové choroby jsou idiopatické (sporadická Creutzfeldova-Jakobova nemoc, CJN), získané - a to iatrogenní (i CJN), nová varianta CJN (nv CJN), kuru - a hereditární (familiární CJN, Gerstmanův-Sträusslerův-Scheinkerův syndrom, GSS, fatální familiární insomnie). Práce je stručný přehled diagnostických kritérií, neuropatologických změn, patogeneze a genetiky lidských prionových chorob. Součástí přehledu jsou první zkušenosti Národní referenční laboratoře přenosných spongiformních encefalopatií a Creutzfeldtovy- Jakobovy nemoci, v níž bylo dosud vyšetřeno 14 případů podezření na lidská prionová onemocnění. Klíčová slova: lidské prionové nemoci, česká Národní referenční laboratoř přenosných encefalopatií a Creutfeldtovy-Jakobovy nemoci Summary The prion diseases are group of neurodegenerative conditions that affect both humans and animals. Human prion diseases are idiopathic (sporadic Creutzfeldt-Jakob disease, CJD), acquired - iatrogenic (i CJD), new variant CJD (nv CJD), kuru, and hereditary (familial CJD, Gerstman-Sträussler-Scheinker syndrome, GSS, fatal familial insomnia). The paper shortly reviews diagnostic criteria, neuropathology, pathogenesis and genetics of human prion diseases. First experiences of the Czech National Reference Laboratory of Transmissible Encephalopathies and Creutzfeldt-Jakob diseases - 14 cases examined are described. Key words: human prion diseases, Czech National Reference Laboratory of Transmissible Encephalopathies and Creutzfeldt-Jakob disease Koukolík F, Matěj R. Lidské prionové nemoci. První zkušenosti a přehled literatury. Psychiatrie 2002;6(3): Prionová onemocnění tvoří skupinu neurodegenerativních chorob postihujících zvířata a lidi. Jejich poznání má dlouhou a složitou historii. Nejstarším známým onemocněním je scrapie (klusavka) postihující ovce, popsána byla roku Nově popsané prionové choroby zvířat jsou encefalopatie norků (1947), chronické kachektizující onemocnění losů a jelenů (CWD, chronic wasting disease, severozápadní státy USA, 1967), kočičí (felinní) spongiformní encefalopatie (FFE, 1990), bovinní spongiformní encefalopatie (BSE, 1986) a stejné onemocnění mnoha desítek zvířecích druhů v evropských zoologických, zahradách včetně bizona a tygra (1986 a dále). Lidská prionová onemocnění jsou sporadická, familiární a iatrogenní Creutzfeldtova-Jakobova nemoc (s CJD, f CJD, i CJD,1920 a dále), Gerstmannova-Sträusslerova-Scheinkerova nemoc, též Gerstmannův-Sträusslerův syndrom (GSS,1936), kuru (1950), fatální familiární insomnie a její sporadická forma (FFI, 1990) a nová varianta CJD (nv CJD, 1996). V roce 1936 byla scrapie přenesena na zdravé ovce a kozy. Nápadná byla dlouhá inkubační doba. Předpokládalo se, že příčinou onemocnění je virus. V roce 1954 užil Sigurdsson pojem infekce pomalými viry. Synonymem se postupně staly pojmy pomalé virózy, přenosné demence a podle typického histologického obrazu spongiformní encefalopatie. V 50. letech 20. století bylo v jazykové skupině Fore, žijící ve Východní vysočině ostrova Papua-Nová Guinea popsáno onemocnění kuru, projevující se progresivní ataxií. Výzkum doložil, že se přenáší rituálním kanibalizmem. V roce 1959 upozornil Hadlow na podobnost kuru a scrapie. Intracerebrální inokulací přenesla kuru na šimpanze roce 1966 Gajdusekova skupina. CJD byla stejným způsobem přenesena Gibbsovou skupinou roku Mastersova skupina přenesla GSS na zvíře v roce Výsledkem této práce byl pojem přenosné demence. Již v roce 1966 se objevily první studie dokazující, že přenosné agens těchto onemocnění nemá nukleové kyseliny. Roku 1967 Griffith dokazoval, že agens může být protein. Boltonova skupina roku 1982 izolovala proteáza-rezistentní sialoglykoprotein, jenž byl hlavní složkou infekční frakce, a označila jej z prionový protein (PrP). Pojem prion vytvořil roku 1982 Prusiner (Prusiner, 1998), označoval jím malé bílkovinné infekční částice, které jsou odolné vůči inaktivaci postupy, které mění nukleové kyseliny. Proteáza rezistentní PrP byl označen PrP27-30 (odpovídá kda). Je kódován genem na 20. chromozomu. Odvozuje se od větší molekuly (33 35 kda) označené PrPSc, protože jde o izoformu proteinu získaného z mozku ovcí postižených scrapie. Normální produkt tohoto genu je vůči účinku proteázy citlivý, označuje se PrPC, což je označení celulární izoformy. Sekvence aminokyselin v řetězci PrPC a PrPSc je stejná. PrPSc vzniká z PrPC posttranslačními procesy (přehled Coolinge, 2001).
, získané - a to iatrogenní (i CJN), nová varianta CJN (nv CJN), kuru - a hereditární (familiární CJN,")
2
3 Lidská prionová onemocnění Sporadická CJN se vyskytuje ve všech zemích světa, kde je možnost tuto chorobu diagnostikovat. Odhadovaný výskyt je 1 případ/milion lidí/rok. V městské populaci a populaci lidí starších než 65 let, jakož i v populaci psychiatrických léčeben může být její výskyt až 5 vyšší. Mezi CJN a scrapie nebyl doložen epidemiologický vztah. Příčina sporadické CJN je neznámá. Předpokládá se možnost spontánní konverze PrP nebo somatické mutace. Přibližně 15 % případů CJN je dědičných. U všech dosud známých případů byly zjištěny mutace genu pro prionový protein (PRNP), v současnosti je jich známo více než 20. V případě bodových mutací je důsledkem substituce jedné aminokyseliny PrP, v jednom případě byl zastižen stop kodon, důsledkem byl rozvětvený PrP. V případě inzercí se kódují společně s pěti kopiemi normálního proteinu do tandemu další kopie oktapeptidových sekvencí (Collinge, 1997). U těchto onemocnění jde o autozomálně dominantní typ dědičnosti. Případy těchto prionových chorob je tedy možné diagnostikovat analýzou PRNP. Ta umožnila odlišení FFI a případné zařazování atypických demencí do rámce prionových nemocí. Genetickým klíčem vnímavosti vůči sporadickým a získaným prionovým chorobám je PRNP polymorfizmus v oblasti kodonu 129, kde je kódován buď metionin, nebo valin. Většina chorob postihuje homozygoty (Windl et al., 1996). Byly popsány rodokmeny, v nichž se vyskytují fenotypy klasických případů CJN a GSS, ale i další neurodegenerativní onemocnění. V některých rodinách se vyskytují jak případy splňující kritéria CJN i GSS, tak atypické případy, které je nesplňují. U těchto atypických prionových chorob se nemusí objevit neurohistologické diagnostické znaky, jako je spongiformní encefalopatie,
, v současnosti je jich známo více než 20.")
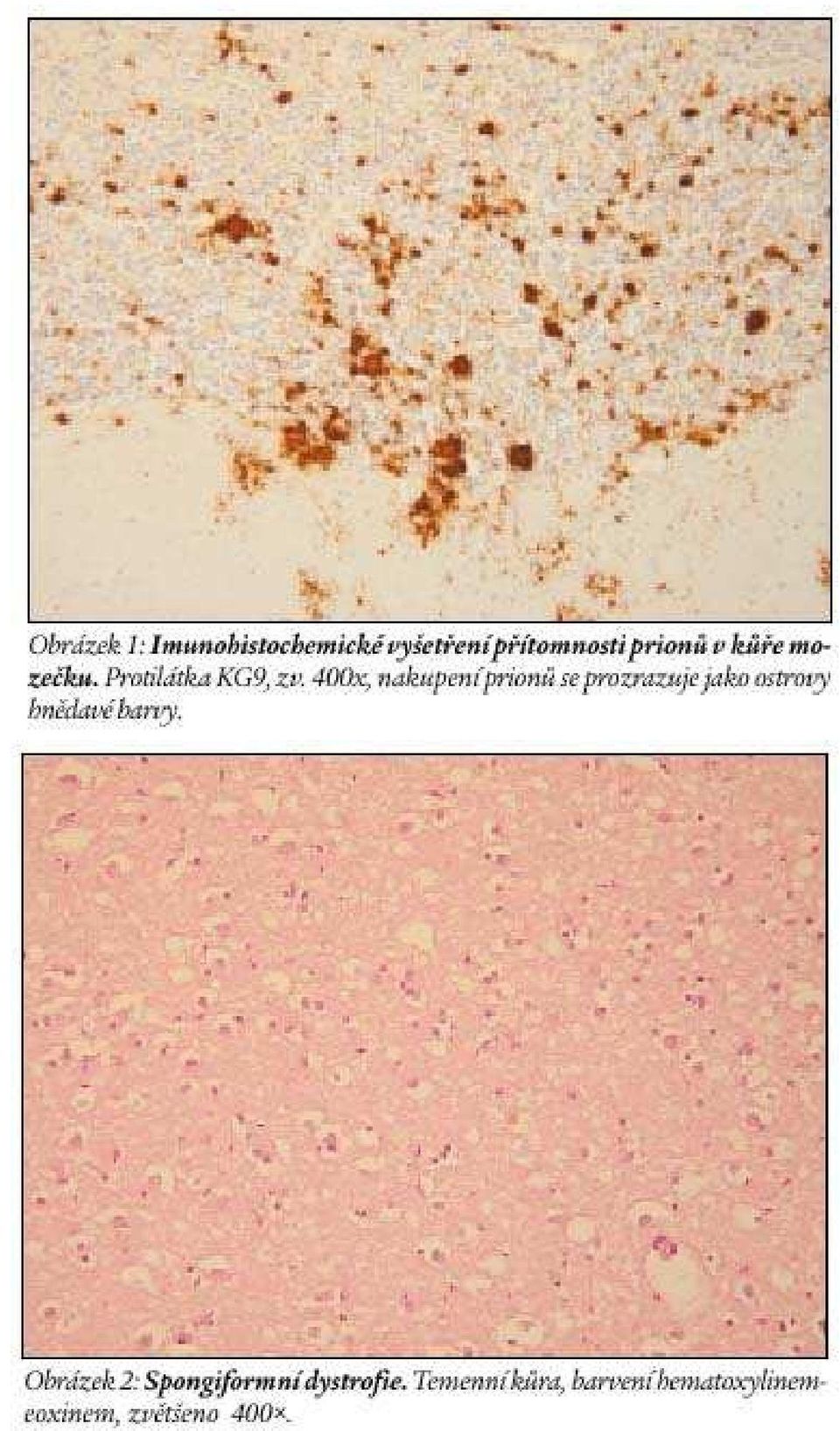
4 imunohistochemické změny však pozitivní bývají. Klinické příznaky těchto atypických prionových chorob zahrnovaly v rozličných kombinacích progresivní demenci, choreu, myoklonus, extrapyramidové příznaky, pseudobulbární příznaky, epileptiformní křeče i amyotrofické změny (Collinge, 2001). Klasická (sporadická) forma CJN se objevuje nejčastěji mezi lety věku (rozmezí let). Obě pohlaví postihuje stejně. Uvádí se výskyt 1 případ/1 milion lidí/rok s tím, že v populaci lidí starších než 65 let a v populaci gerontopsychiatrických oddělení by mohl být vyšší. Prodromální příznaky postihují asi třetinu nemocných a jsou nespecifické: únava, nespavost, deprese, ztráta na váze, bolesti hlavy, celková nevolnost, špatně definovatelné bolesti. V průběhu měsíců se vyvíjí mentální deteriorace do hluboké demence, případně akinetického mutizmu. Vývoj příznaků demence doprovází myoklonus, extrapyramidové a pyramidové příznaky, cerebelární ataxie, případně korová slepota. Myoklonus je velmi rychlý svalový záškub s motorickým efektem, častý je ve stejných svalových skupinách. Opakuje se rytmicky, nebo nepravidelně. Typický je EEG nález generalizované difázické nebo trifázické vlny, které se periodicky opakují v intervalech 0,5 2 s, trvají s. Tyto změny se v případech s CJN vyvíjejí postupně, proto je nutné zhotovit často řadu opakovaných záznamů. Kromě toho se nevyskytují po celou dobu onemocnění. Někdy nejsou přítomny vůbec. K diagnóze napomáhá zjištění proteinu v mozkomíšním moku (Green et al., 2001). Onemocnění probíhá nejčastěji několik měsíců, ve většině případů dobu kratší než jeden rok. Diferenciální diagnóza CJN je diferenciální diagnózou syndromu demence (Koukolík a Jirák, 1999). Současná klasifikace CJN je v tabulce 1. Definitivní diagnóza CJN (stejně jako dalších prionových onemocnění) je neurohistologická a imunohistochemická (viz níže), nutné je i vyšetření westernblotem a molekulárně genetické. Iatrogenní CJN (v současnosti se užívá pojem náhodně přenesená, accidentally transmitted CJN) byla způsobena transplantací rohovky, užitím nedostatečně sterilizovaných intracerebrálních elektrod, transplantací dura mater, dále užitím růstového hormonu a gonadotrofinu získaného z kadaverózních hypofýz (Prusiner, 2001; Report, 1997). Případy, které byly důsledkem intracerebrální nebo intraokulární inokulace, jsou klinicky blízké klasické CJN s rychle progedující demencí. Případy dané periferní inokulací se projevují více jako cerebelární ataxie připomínajicí kuru, o které je níže. Inkubace po transplatanci dura mater se pohybovala mezi měsíci, inkubace po periferní inokulaci byla v průměru 15letá. Familiární podoba CJN je způsobena v mutacemi PRNP. Diagnostikuje se na základě neuropsychiatrických příznaků, z nichž lze vyvodit pravděpodobnou, možnou, případně definitivní diagnózu CJN, dále na základě pravděpodobné, možné nebo definitivní diagnózy CJN u příbuzného v 1. stupni příbuzenství a na základě zjištění mutace PRNP charakteristické pro takové onemocnění. Rodokmeny postižené těmito onemocněními se vyskytují v oblasti Kysuce a na jižním Slovensku s přesahem do přilehlé oblasti Maďarska (Mitrová, 1999). Nové případy familiárních prionových onemocnění přibývají (viz např. Kovács et al., 2001). V rodokmenech postižených familiární podobou CJN se mohou objevit výše zmíněné atypické případy prionových chorob. Nová varianta CJN (nvcjn, též vcjn) byla popsána na 10 případech v roce 1996 (Will et al., 1996), které byly výsledkem epidemiologického dohledu nad výskytem Creutzfeldtovy-Jakobovy nemoci ve Spojeném království, zavedeném v roce Smyslem dozoru bylo ověřit výskyt této nemoci ve vztahu k epidemii bovinní spongiformní encefalopatie (BSE), která v této zemi začala v roce Do roku 1996 bylo v této laboratoři neuropatologicky vyšetřeno 207 případů CJN. Iatrogenní a familiární formy onemocnění byly ze sestavy vyloučeny. V souboru bylo zjištěno 10 zemřelých ve věkovém rozmezí let (medián 29). Interval mezi prvními příznaky a úmrtím byl 7,5 22,5 měsíců (medián 12). U 185 ostatních případů CJD byl střední věk začátku choroby 65 let, střední průběh 4 měsíce. Uvedených 10 případů mělo klinické příznaky, které se odlišovaly od klasické CJN. U čtyř se vyskytovaly dysestezie, u jednoho bolesti v oblasti nohy, u 9 změny chování, pro které byli odesláni k psychiatrickému vyšetření. U 9 byla raným příznakem mozečková ataxie. Poruchy paměti se na počátku choroby objevily jen u 2, u všech se však postupně projevovala progresivní demence. Sedm pacientů postihl myoklonus. U žádného nebyly zjištěny změny EEG charakteristické pro CJN. V současnosti je těchto zemřelých popsáno více než 100. Téměř všichni pocházejí ze Spojeného království, 3 případy, které nebyly v této zemi, byly popsány ve Francii, 1 podezření bylo vysloveno ve Španělsku a zcela nově 1 podezření v Itálii. V případě podezření na tuto chorobu je možné odebrat pacientům tonsilu, případně appendix, a v jejich lymforetikulární tkáni určit přítomnost prionů imunohistochemicky. Gerstmannova-Sträusslerova-Scheinkerova nemoc (GSS) je autozomálně dominantní onemocnění, které se klinicky projevuje mozečkovou ataxií s pyramidovými příznaky, později nastupuje demence, která nemusí být hluboká. Kromě těchto příznaků se objevuje dysfagie, dysartrie, hyporeflexie. Průběh onemocnění je obvykle pětiletý, podstatně tedy delší než u CJN. První příznaky se objevují ve třetí nebo čtvrté dekádě života. Jde o syndrom, jenž je důsledkem několika druhů mutací PRNP. Fatální familiární insomnie (FFI) se projevuje nespavostí a dysautonomií, později se objeví ataxie, dysartrie, myoklonus a pyramidové příznaky. V závěru onemocnění je insomnie úplná, doprovází ji demence, rigidita, dystonie a mutizmus. Příčinou je mutace D178N PRNP asociovaná s metioninovým kodonem 129 na stejné alele (Medori et al., 1992). Kuru postihuje obě pohlaví, častěji však ženy a děti. Nejmladší postižené děti byly pětileté, nejstarším nemocným bylo více než 60 let. Onemocnění trvá v průměru 12 měsíců (3 měsíce až 3 roky). Klíčovým příznakem je progresivní mozečková ataxie (slovo kuru označuje v místním jazyce třes). Demence se obvykle nevyvíjí. Předpokládá se, že pramenem kuru byl případ sporadické CJN na počátku 20. století. Inkubační doba kuru je nejčastěji kolem 12 let, může přesáhnout 40 let (Klitzman et al., 1984; Prusiner a McKinley, 1987). Definitivní diagnóza lidských prionových chorob Definitivní diagnóza lidských prionových chorob je založena na neurohistologickém vyšetření doplněném imunohistochemickými metodami a metodou westernblot, které ověřují přítomnost prionů ve tkáni. Klasickou neurohistologickou trojicí změn při prionových nemocech je spongiformní dystrofie, numerická atrofie neuronů a glióza. Spongiformní dystrofii charakterizují vakuoly v neuropilu jejichž průměr je 2 20 m. Objevují se ložiskově, je nutné odlišit je od artefaktů a od vakuol, které se objevují v horních korových vrstvách při různých příčinách korové atrofie. Imunohistochemické vyšetření užívá různé typy protilátek ozřejmujících výskyt prionů ve tkáni (obr. 1, 2). Vysoce charakteristické je, že priony ve tkáni nevyvolávají zánětlivou odpověď, což byl jeden z důvodů dlouhého tápání při objasňování povahy této nemoci. Nález při nv CJN se od změn při s CJN odlišuje histologicky i imunohistochemicky. Novou variantu CJN charakterizuje velký počet floridních plak (obr. 3) v kůře mozku i mozečku, shluky mlaých plak v imunohistochemickém obraze, amorfní

5 pericelulární a perivaskulární kupení PrP, těžké spogniformní změny a perineuronální a axonální kupení PrP v neostriatu, výrazná astrocytóza a numerická atrofie neuronů v zadních talamických jádrech a mezencefalu, perineurální a retikulární kupení PrP v šedé hmotě kmene a spinální míchy. Kromě toho odlišuje nv CJN od s CJN i analýza westernblotu (Ironside et al., 2000). Genetika lidských prionových chorob Lidský prionový gen (PRNP) je na krátkém raménku chromozomu 20. Sestává ze dvou exonů a jednoho intronu. Celá kódující oblast (čtecí rámeček) je v oblasti exonu 2. Zde se zjišťují bodové mutace a inzerce v rodinách postižených dědičnými prionovými nemocemi. (Exon je část genu, jejíž odpovídající sekvence je přítomna v odpovídající mrnk. Intron odpovídá části genu, která se transkribuje, nicméně je při tvorbě mrnk z primárního transkriptu odstraněna procesem nazývaným splicing, takže zůstává v jádře. Inzerce je druh mutace podmíněný vložením dalších bází do genu). Mutace P102L je nejčastější příčinou GSS, byla zjištěna v rodině původně popsané těmito autory. Tkáň z případů P102L byla užita k přenosu choroby na hlodavce a primáty. Zdařený přenos dokazuje infekční povahu onemocnění. Mutace D178N-CJN/FFI Valin kódovaný kodonem 129 mutované alely D178N se vyskytuje u familiární CJN. Metionin kódovaný kodonem 129 společně s mutací D178N je příčinou FFI. E200K je v rámci dědičných lidských prionových chorob nejrozšířenější mutace, klinicky i neuropatologicky se velmi podobá CJN. Nejvyšší výskyt je znám mezi lybijskými Židy v Izraeli, kde je incidence CJN 100 vyšší než u ostatní světové populace. Existuje řada dalších vzácných mutací zjištěných v malém počtu rodin, kde se vyskytly příznaky CJN, GSS nebo atypických prionových chorob. Odlišný typ genetické změny představují rozlišné druhy inzercí. Normální gen obsahuje 5 opakujících se sekvencí (1 devítičlennou, 4 osmičlenné), při inzercích se objevuje 1 9 dalších osmičlenných opakujících se sekvencí. Patogeneticky klíčový je polymorfizmus v oblasti kodonu 129, kde sekvence ATG kóduje metionin a sekvence GIT valin. V souborech scjn je v porovnání se zdravou populací daleko vyšší výskyt homozygotů pro metionin. Jimi jsou i všichni dosud zjištění pacienti s nvcjn. Prionové proteiny Lidský PrPc je membránový glykoprotein exprimovaný v neuronech, astrocytech a řadě dalších buněčných druhů. Zralý PrPc je v membráně ukotven svým glykosylovaným zakončením. Strukturální studie magnetickou rezonancí ukazují, že C-terminální polovina molekuly obsahuje dva antiparalelní beta-plizované úseky (S1,S2, tvar připomíná plizování sukně) a tři alfa helikální oblasti (H1 H3, tvar připomíná péro v matraci). Funkce PrPc je nejasná. Uvádí se vztah k vazbě mědi a synaptickému přenosu (Brown et al., 1997). PrP se axonálním transportem dostává do synapsí jak v případě normálních mozků, tak v mozcích infikovaných scrapie. Konverze PrPc na PrPSc, která se považuje za klíčovou patogenetickou událost ve vývoji prionových nemocí, pravděpodobně probíhá na buněčném povrchu nebo po proběhlé endocytóze. Kinetika této konverze je nejasná. PrPSc se odlišuje podstatně větším podílem beta-plizovaných oblastí. Aminokyselinové složení obou molekul zůstává stejné, normální od patologického PrP se tedy liší podle současného poznání jen prostorovým uspořádáním. Předpokládá se, že různé typy odlišného prostorového uspořádání a glykosylace jsou podkladem existence různých prionových kmenů, resp. typů (Collinge et al., 1996). Příčinou sporadické a iatrogenní formy CJN jsou typy 1 3, všechny dosud zjištěné případy nv CJN zapříčinil odlišný typ 4 (Collinge et al., 1996; Wadsworth et al., 1999). Z myšího modelu scrapie plyne, že mechanizmus zániku neuronů při prionových infekcích je apoptóza (Giese et al., 1998). Jejím podkladem by mohlo být poškození synaptického přenosu a/nebo toxický vliv patologického prionu. Nová varianta CJN a bovinní spongiformní encefalopatie V roce 1986 byla ve Spojeném království zjištěna BSE. Předpokládalo se, že je důsledkem přenosu scrapie ovcí, která je v této zemi endemická, na hovězí dobytek, a to kontaminovanou potravou. Multifokální výskyt BSE svědčil pro široce rozšířené agens. Analýza kmene, jenž je příčinou BSE, však ukázala, že se odlišuje od kmene, jenž je rozšířenou příčinou scrapie. Buď jde tedy o vzácný kmen scrapie, nebo o možnost, že se do krmiva pro hovězí dobytek dostal případ sporadické BSE, k čemuž mohlo dojít někdy počátkem 80. let minulého století, jak dokazuje Phillipsova zpráva (Lancet, 2000). Stejně jako těla uhynulých ovcí se totiž na masokostní moučku zpracovávají i těla uhynulého hovězího dobytka. Ve stejné době se začaly objevovat první případy felinní spongiformní encefalopatie a stejného postižení u řady druhů zvířat v zoologických zahradách. Ve vyšetřovaných případech těchto onemocnění byly zjištěny prionové kmemy označené BSE-like (Bruce et al., 1994). Nová varianta CJN byla diagnostikována ve Spojeném království v roce 1996 (Will et al., 1996). Vztah nv CJN k BSE určila typizace kmene prionů (Collinge a kol., 1996) a experimentální přenos na transgenní i konvenční myš (Hill et al., 1997; Bruce et al., 1997). Z těchto studií plyne, že nv CJN je s vysokou pravděpodobností lidská BSE (Collinge, 1999). Předpokládá se, že přenos prionů BSE na lidi je alimentární. Odhaduje se, že do lidského potravního řetězce se dostalo před rokem 1986 asi kusů infikovaných zvířat, podstatně větší počet poté. Zákaz užívání rizikových součástí z porážek byl ve Spojeném království vysloven roku Pravděpodobnost přenosu záleží na individuální dávce, genetické vnímavosti a výši mezidruhové bariéry mezi hovězím dobytkem a člověkem. Předpokládá se, že ta je závislá na odlišných genotypech PRNP. Podle dosavadních zjištění jsou vnímaví jen homozygoti met-met na 129 kodonu PRNP, což je asi 37 % dosud vyšetřené populace (zbytek jsou heterozygoti met-val nebo homozygoti val-val). Není však jisté, zda nejde o špičku ledovce, která se projevuje kratší inkubační dobou. Mechanizmus, jímž se priony dostávají do CNS, je nejasný. Primární cílovou tkání při alimentárním přenosu by mohla být lymforetikulární tkáň zažívací trubice a sleziny. Dostupná data nedovolují určit rozsah možné epidemie odhady se pohybují od několika set případů do více než případů v příštích 30 letech (Balter, 2001; Valleron a kol., 2001; Huillard d Aignaux, 2001). Nově byl diagnostikován případ nvcjn u 74 roků starého muže (Lorains a kol., 2001), čímž padla dosud odhadovaná věková bariéra. S ohledem na mimořádnou odolnost prionů vůčí běžným dezinfekčním prostředkům, dlouhou inkubační dobu, pravděpodobnou infektivitu již v průběhu inkubace, jsou prionová onemocnění těžkým epidemiologickým problémem všech operačních oborů i transplantační medicíny, což dokázal nedávný případ britského pacienta Tony Barretta, jemuž byla provedena apendektomie. Nástroje byly sterilizovány obvyklým způsobem, jenž ponechává priony

6 nedotčené. Šest měsíců poté u pacienta propukly příznaky nv CJN (Dyer, 2001). Národní referenční laboratoř transmisivních spongiformních encefalopatií a Creuzfeldtovy-Jakobovy nemoci ČR Laboratoř byla vybudována z rozhodnutí ministra zdravotnictví ČR při oddělení patologie Fakultní Thomayerovy nemocnice. Současně byla pro klinickou diagnostiku prionových onemocnění vyčleněna lůžka na zdejším neurologickém oddělení (ved. prim. dr. P. Bočan, CSc.) Činnost zahájila laboratoř Je přístrojově plně vybavena. Soustředí se na diferenciální diagnostiku neurodegenerativních onemocnění s cílem vyhledávat případy prionových onemocnění. Základním smyslem činnosti je dohled nad možným výskytem nv CJN. Zavedeny jsou neurohistologické a imunohistochemické metody. Western blot a molekulárně genetické vyšetřování popis kodonu 129 prionového genu se zavádí ve spolupráci s imunoložkou a genetičkou z příslušných laboratoří naší nemocnice. (Westernblot, metoda, které se také říká imunoblot, je imunologická metoda, jejímž principem je elektroforetické rozdělení proteinů na membráně a jejich detekce příslušnými antilátkami). Laboratoř spolupracuje s referenční laboratoří prionových nemocí WHO, která je ve skotském Edinburghu. Všechna oddělení patologie ČR byla písemně informována o způsobu spolupráce v případě podezření na lidské prionové onemocnění. První zkušenosti laboratoře shrnuje tabulka 2. MUDr. František Koukolík, DrSc. Národní referenční laboratoř TSE/CJN Oddělení patologie Fakultní Thomayerovy nemocnice s poliklinikou v Praze-Krči Vídeňská Praha 4 Do redakce došlo: 25. února 2002 Přijato k publikaci: 1. srpna 2002 Literatura Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser P, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzchmar H. The celullar prion protein binds copper in vivo. Nature 1997;390: Bruce M, Chree A, McConell I, Foster J, Pearson G, Fraser H. Trans-mission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci 1994;343: Bruce ME, Will RG, Ironside JW. Transmission to mice indicate that the new variant CJD is caused by the BSE agent. Nature 1997; 389: Collinge J, Siddle KCL, Meads J, Ironside, J, Hill, AF. Molecular analysis of prion strain variation and the aetiology of new variant CJD. Nature 1996;383: Collinge J. Human prion diseases and bovine spongiform encephalo-pathy (BSE). Hum Mol Genet 1997;6: Collinge J. Variant Creutzfeldt-Jakob disease. Lancet 1999;354: Collinge J. Prion diseases of human and animals: their causes and molecular basis. Annu Rev Neurosci 2001;24: Dyer O. Hospitals to spend L 200m to prevent spread of v CJD. Brit Med J 2001;322:68. Giese A, Brown DR, Groschup M, Feldman C, Haist I, Kretschmar HA. Role of microglia in neuronal death in prion disease. Brain Pathol 1998;8: Green AJE, Thompson EJ, Stewart GE, Zeidler M, Mc Kenzie JM, MacLeod M-A, Ironside JW, Will RG, Knight RSG. Use of and other brain-specific proteins in CSF in the diagnosis of variant Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2001;70: Hill AF, Desbruslais M, Joiner S, Sidle KCL, Gowland I, Collinge J. The same prion strain causes vcjd and BSE. Nature 1997;389: Huillard d Aignaux JN, Cousens SN, Smith PG. Predictability of the UK variant Creutzfeldt-Jakob disease epidemic. Science 2001;294: Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt-Jakob disease. Histopathology 2000; 37:1 9. Klitzman RL, Alpers MP, Gajdusek DC. The natural incubation of kuru and the episodes of transmission in thress clusters of patients. Neuroepidemiology 1984;3:3 20. Koukolík F, Jirák R. Diagnostika a léčení syndromu demence. Praha: Grada Publishing, Kovács GG, Ertsey C, Majtényi C, Jelencsik I, László L, Flicker H, Strain L, Szirmai I, Budka H. Inherited prion disease with A117V mutation of the prion protein gene: a novel hungarian family. J Neurol Neurosurg Psychiatry 2001;70: Lancet editorial. The Phillips report on BSE and vcjd. Lancet 2001; 356:1535. Lorains JW, Henry C Agbamu DA, Rossi M, Bishop M, Will RG, Ironside JW. Variant Creutzfeldt-Jakob disease in an elderly patient. Lancet 2001; 357: Medley, GF. Predicting the unpredictable. Science 2001;294: Medori R, Tritschler JH, LeBlanc A. Fatal familial insomnia a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med 1992;326: Mitrová E. Transmissibilné spongioformné encefalopátie. Prionové choroby. Press Publica, Prusiner SB, McKinley MP, eds. Prions: Novel Infectious Pathogens Causing Scrapie and Creutzfeldt-Jakob Disease. San Diego: Academic Press, 1987; Prusiner SB. Prions. Proc Natl Acad Sci USA 1998;95: Prusiner SB. Shattuck lecture - neurodegenerative diseases and prions. New Engl J Med 2001;344: Report on human growth hormone and Creutzfeldt-Jakob disease. Washington D.C.: Public Health Service Interagency
 Činnost zahájila laboratoř 1. 7. 2001. Je přístrojově plně vybavena. Soustředí se na diferenciální diagnostiku neurodegenerativních onemocnění s cílem vyhledávat případy prionových onemocnění.")
7 Coordinating Comittee, 1997;14:1 11. Valleron A-J, Boelle P-Y, Will R, Cesbron J-Y. estimation of epidemic size and incubation time based on age characteristics of vcjd in the United Kingdom. Science 2001;294: Wadsworth JDF, Hill AF, Joiner, S, Jackson GS, Clarke AR, Collinge J. Strain-specific prion-protein conformation determined by metal ions. Nat Cell Biol 1999;1: Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfedlt-Jakob disease in UK. Lancet 1996;347: Windl O, Dempster M, Estibeiro JP, Lathe R, De Silva R, Esmonde T, Will R, Springbett A, Campbell TA, Sidle KC, Palmer MS, Collinge J. Genetic basis of Creutzfeldt-Jakob disease in the United Kingdom: a systematic analysis of predisposing mutations and allelica varitation in PRNP gene. Hum Genet 1996;98: Left.gif (10785 Midlle.gif (12243 Right.gif (10788

Lidská prionová onemocnění Radoslav Matěj, Robert Rusina
 Lidská prionová onemocnění Radoslav Matěj, Robert Rusina Oddělení patologie a molekulární medicíny Neurologické oddělení Thomayerova nemocnice Transmissible Spongiform Encephalopathy Terminologie Priony
Lidská prionová onemocnění Radoslav Matěj, Robert Rusina Oddělení patologie a molekulární medicíny Neurologické oddělení Thomayerova nemocnice Transmissible Spongiform Encephalopathy Terminologie Priony
B S E. MUDr. Miroslava Zavřelová. Ústav preventivního lékařství LF MU
 B S E MUDr. Miroslava Zavřelová Ústav preventivního lékařství LF MU TSE / BSE TSE transmisibilní spongiformní encefalopatie BSE bovinní spongiformní enecefalopatie Prionové nákazy zvířat Scrapie onemocnění
B S E MUDr. Miroslava Zavřelová Ústav preventivního lékařství LF MU TSE / BSE TSE transmisibilní spongiformní encefalopatie BSE bovinní spongiformní enecefalopatie Prionové nákazy zvířat Scrapie onemocnění
Diagnostika prionů. Vladislava Růžičková. XVI. ročník kurzu pro učitele 2014, Brno
 Diagnostika prionů Vladislava Růžičková XVI. ročník kurzu pro učitele 2014, Brno Diagnostika prionů Prion (PrP Sc -patologická forma prionového proteinu (PrP C ). Stanley Prusiner - 1997 Nobel Prize Vyznačují
Diagnostika prionů Vladislava Růžičková XVI. ročník kurzu pro učitele 2014, Brno Diagnostika prionů Prion (PrP Sc -patologická forma prionového proteinu (PrP C ). Stanley Prusiner - 1997 Nobel Prize Vyznačují
Neurodegenerativní onemocnění
 Neurodegenerativní onemocnění Radoslav Matěj Oddělení patologie a molekulární medicíny TN Centrum pro diagnostiku a studium neurodegenerativních onemocnění Neurodegenerativní onemocnění Úbytek neuronálních
Neurodegenerativní onemocnění Radoslav Matěj Oddělení patologie a molekulární medicíny TN Centrum pro diagnostiku a studium neurodegenerativních onemocnění Neurodegenerativní onemocnění Úbytek neuronálních
PŘEHLEDNÝ REFERÁT. R. Matěj 1, R. Rusina 2, F. Koukolík 1
 PŘEHLEDNÝ REFERÁT 5 let činnosti Národní referenční laboratoře lidských prionových onemocnění při Oddělení patologie a molekulární medicíny FTNsP: naše zkušenosti a přehled literatury 5 Years of Activities
PŘEHLEDNÝ REFERÁT 5 let činnosti Národní referenční laboratoře lidských prionových onemocnění při Oddělení patologie a molekulární medicíny FTNsP: naše zkušenosti a přehled literatury 5 Years of Activities
Diagnostika prionů 2014
 Předmět: Bi 8360 Molekulární diagnostika mikroorganismů Diagnostika prionů 2014 Vladislava Růžičková Prion "proteinaceous infectious particle proteinová infekční částice Prion (PrP Sc -patologická forma
Předmět: Bi 8360 Molekulární diagnostika mikroorganismů Diagnostika prionů 2014 Vladislava Růžičková Prion "proteinaceous infectious particle proteinová infekční částice Prion (PrP Sc -patologická forma
Neurologická klinika IPVZ a Thomayerovy nemocnice, Národní referenční laboratoř pro diagnostiku lidských prionových nemocí, Praha 2
 78 Prionová onemocnění MUDr. Robert Rusina, Ph.D. 1, MUDr. Radoslav Matěj, Ph.D. 2 1 Neurologická klinika IPVZ a Thomayerovy nemocnice, Národní referenční laboratoř pro diagnostiku lidských prionových
78 Prionová onemocnění MUDr. Robert Rusina, Ph.D. 1, MUDr. Radoslav Matěj, Ph.D. 2 1 Neurologická klinika IPVZ a Thomayerovy nemocnice, Národní referenční laboratoř pro diagnostiku lidských prionových
Závěrečné zhodnocení iatrogenního přenosu nemoci Creutzfeldta-Jakoba (Iatrogenic Creutzfeldt-Jakob Disease, Final Assessment)
") 1 Studijní materiál speciál č. 146 Srpen 2012 Závěrečné zhodnocení iatrogenního přenosu nemoci Creutzfeldta-Jakoba (Iatrogenic Creutzfeldt-Jakob Disease, Final Assessment) Brown P., Brandel J-Ph., Sato
1 Studijní materiál speciál č. 146 Srpen 2012 Závěrečné zhodnocení iatrogenního přenosu nemoci Creutzfeldta-Jakoba (Iatrogenic Creutzfeldt-Jakob Disease, Final Assessment) Brown P., Brandel J-Ph., Sato
B S E MUDr. Miroslava Zavřelová Mgr. Aleš Peřina, Ph. D. Ústav preventivního lékařství LF MU
 Snímek 1 B S E MUDr. Miroslava Zavřelová Mgr. Aleš Peřina, Ph. D. Ústav preventivního lékařství LF MU Snímek 2 TSE u zvířat Scrapie (klusavka, drbavka) onemocnění ovcí a koz 1738: Velká Británie, Francie
Snímek 1 B S E MUDr. Miroslava Zavřelová Mgr. Aleš Peřina, Ph. D. Ústav preventivního lékařství LF MU Snímek 2 TSE u zvířat Scrapie (klusavka, drbavka) onemocnění ovcí a koz 1738: Velká Británie, Francie
Nová varianta CJD (Variant Creutzfeldt-Jakob disease) Collinge J. Lancet, Vol. 354, 1999, č. 9175, s. 317-323 Volně přeložil a zkrátil MUDr.
 Collinge J. Lancet, Vol. 354, 1999, č. 9175, s. 317-323 Volně přeložil a zkrátil MUDr.") Studijní materiál důchodce č. 11 Srpen 1999 Nová varianta CJD (Variant Creutzfeldt-Jakob disease) Collinge J. Lancet, Vol. 354, 1999, č. 9175, s. 317-323 Volně přeložil a zkrátil MUDr. Plesník Souhrn Je
Studijní materiál důchodce č. 11 Srpen 1999 Nová varianta CJD (Variant Creutzfeldt-Jakob disease) Collinge J. Lancet, Vol. 354, 1999, č. 9175, s. 317-323 Volně přeložil a zkrátil MUDr. Plesník Souhrn Je
Fond rozvoje vysokých škol okruh G studentské projekty
 Fond rozvoje vysokých škol okruh G studentské projekty EXCELENCE DOKTORSKÉHO STUDIA NA AF MENDELU PRO NAVAZUJÍCÍ EVROPSKOU VĚDECKO-VÝZKUMNOU KARIÉRU CZ.1.07/2.3.00/20.0005 Tento seminář je spolufinancován
Fond rozvoje vysokých škol okruh G studentské projekty EXCELENCE DOKTORSKÉHO STUDIA NA AF MENDELU PRO NAVAZUJÍCÍ EVROPSKOU VĚDECKO-VÝZKUMNOU KARIÉRU CZ.1.07/2.3.00/20.0005 Tento seminář je spolufinancován
Huntingtonova choroba
 Huntingtonova choroba Renata Gaillyová OLG FN Brno Huntingtonova choroba je dědičné neurodegenerativní onemocnění mozku, které postihuje jedince obojího pohlaví příznaky se obvykle začínají objevovat mezi
Huntingtonova choroba Renata Gaillyová OLG FN Brno Huntingtonova choroba je dědičné neurodegenerativní onemocnění mozku, které postihuje jedince obojího pohlaví příznaky se obvykle začínají objevovat mezi
Lidská prionová onemocnění
 Univerzita Karlova v Praze Farmaceutická fakulta v Hradci Králové Katedra biologických a lékařských věd Lidská prionová onemocnění Bakalářská práce Praha, 2007 Eva Parobková Lidská prionová onemocnění
Univerzita Karlova v Praze Farmaceutická fakulta v Hradci Králové Katedra biologických a lékařských věd Lidská prionová onemocnění Bakalářská práce Praha, 2007 Eva Parobková Lidská prionová onemocnění
Patologie nervového systému. XI. histologické praktikum 3. ročník všeobecného směru
 Patologie nervového systému XI. histologické praktikum 3. ročník všeobecného směru Malacie mozková Malacie mozková Hemoragie mozková Hemoragie mozková Subarachnoideální krvácení Hnisavá leptomeningitis
Patologie nervového systému XI. histologické praktikum 3. ročník všeobecného směru Malacie mozková Malacie mozková Hemoragie mozková Hemoragie mozková Subarachnoideální krvácení Hnisavá leptomeningitis
Obsah. 1. Gerontopsychiatrie - historie, osobnosti, současnost (Roman Jirák) 2. Nejčastější psychické poruchy v seniorském věku (Roman Jirák)
 2. Nejčastější psychické poruchy v seniorském věku (Roman Jirák)") Obsah 1. Gerontopsychiatrie - historie, osobnosti, současnost (Roman Jirák) 2. Nejčastější psychické poruchy v seniorském věku (Roman Jirák) 3. Změny psychiky ve stáří (Tamara Tošnerová) Ztráta nezávislosti
Obsah 1. Gerontopsychiatrie - historie, osobnosti, současnost (Roman Jirák) 2. Nejčastější psychické poruchy v seniorském věku (Roman Jirák) 3. Změny psychiky ve stáří (Tamara Tošnerová) Ztráta nezávislosti
Elektronoptický snímek viru mozaikové choroby tabáku. Mozaiková choroba tabáku. Schéma viru mozaikové choroby tabáku
 Obecná virologie Viry lat. virus šťáva, jed, v lékařské terminologii infekční činitel 1879 1882: první pokusný přenos virového onemocnění (mozaiková choroba tabáku) 1898: první pokusný přenos živočišného
Obecná virologie Viry lat. virus šťáva, jed, v lékařské terminologii infekční činitel 1879 1882: první pokusný přenos virového onemocnění (mozaiková choroba tabáku) 1898: první pokusný přenos živočišného
Epidemiologie. MUDr. Miroslava Zavřelová Ústav ochrany a podpory zdraví LF MU
 Epidemiologie MUDr. Miroslava Zavřelová Ústav ochrany a podpory zdraví LF MU Epidemiologie Studium hromadně se vyskytujících jevů Stanovení opatření intervence Analýza efektivity intervence Epidemiologie
Epidemiologie MUDr. Miroslava Zavřelová Ústav ochrany a podpory zdraví LF MU Epidemiologie Studium hromadně se vyskytujících jevů Stanovení opatření intervence Analýza efektivity intervence Epidemiologie
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354
 I n v e s t i c e d o r o z v o j e v z d ě l á v á n í Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním
I n v e s t i c e d o r o z v o j e v z d ě l á v á n í Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním
Inovace studia molekulární a buněčné biologie
 Inovace studia molekulární a buněčné biologie Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky. OBVSB/Obecná virologie Tento projekt je spolufinancován Evropským
Inovace studia molekulární a buněčné biologie Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky. OBVSB/Obecná virologie Tento projekt je spolufinancován Evropským
Nová hypotéza o původu BSE (The origin of bovine spongiform encaphalopathy: the human prion disease hypothesis)
") 1 Studijní materiál speciál č. 17 Květen 2006 Psáno na počest 75. narozenin dvou milých přátel MUDr. Bartoloměje Bindase a MUDr. Witolda Gawlase Nová hypotéza o původu BSE (The origin of bovine spongiform
1 Studijní materiál speciál č. 17 Květen 2006 Psáno na počest 75. narozenin dvou milých přátel MUDr. Bartoloměje Bindase a MUDr. Witolda Gawlase Nová hypotéza o původu BSE (The origin of bovine spongiform
CADASIL. H. Vlášková, M. Boučková Hnízdová, A. Loužecká, M. Hřebíček, R. Matěj, M. Elleder
 CADASIL analýza mutací v genu NOTCH3 H. Vlášková, M. Boučková Hnízdová, A. Loužecká, M. Hřebíček, R. Matěj, M. Elleder Ústav dědičných metabolických poruch 1. LF UK a VFN Oddělení patologie a nár. ref.
CADASIL analýza mutací v genu NOTCH3 H. Vlášková, M. Boučková Hnízdová, A. Loužecká, M. Hřebíček, R. Matěj, M. Elleder Ústav dědičných metabolických poruch 1. LF UK a VFN Oddělení patologie a nár. ref.
BSE na počátku třetího tisíciletí Zprac.: MUDr. Vladimír Plesník
 1 Novoroční speciál Studijní materiál důchodce č. 146 Leden 2003 BSE na počátku třetího tisíciletí Zprac.: MUDr. Vladimír Plesník 1. Situace ve Velké Británii Úředně bylo ve VB potvrzeno k 1. 5. 2001 celkem
1 Novoroční speciál Studijní materiál důchodce č. 146 Leden 2003 BSE na počátku třetího tisíciletí Zprac.: MUDr. Vladimír Plesník 1. Situace ve Velké Británii Úředně bylo ve VB potvrzeno k 1. 5. 2001 celkem
Výskyt rotavirových onemocnění v České republice v letech , EpiDat Michaela Špačková, Martin Gašpárek
 Výskyt rotavirových onemocnění v České republice v letech 1997-2017, EpiDat Michaela Špačková, Martin Gašpárek Oddělení epidemiologie infekčních nemocí, Národní referenční centrum pro analýzu epidemiologických
Výskyt rotavirových onemocnění v České republice v letech 1997-2017, EpiDat Michaela Špačková, Martin Gašpárek Oddělení epidemiologie infekčních nemocí, Národní referenční centrum pro analýzu epidemiologických
Péče o pacienty léčené pro demence v ambulantních a lůžkových zařízeních ČR v letech
 Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 31. 12. 2012 66 Péče o pacienty léčené pro demence v ambulantních a lůžkových zařízeních ČR v letech 2007 2011 Health
Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 31. 12. 2012 66 Péče o pacienty léčené pro demence v ambulantních a lůžkových zařízeních ČR v letech 2007 2011 Health
Potřebné genetické testy pro výzkum a jejich dostupnost, spolupráce s neurology Taťána Maříková. Parent projekt. Praha 19.2.2009
 Potřebné genetické testy pro výzkum a jejich dostupnost, spolupráce s neurology Taťána Maříková Parent projekt Praha 19.2.2009 Diagnostika MD její vývoj 1981-1986: zdokonalování diferenciální diagnostiky
Potřebné genetické testy pro výzkum a jejich dostupnost, spolupráce s neurology Taťána Maříková Parent projekt Praha 19.2.2009 Diagnostika MD její vývoj 1981-1986: zdokonalování diferenciální diagnostiky
Registrační číslo projektu: CZ.1.07/1.5.00/34.0649. Základy genetiky - geneticky podmíněné nemoci
 Výukový materiál zpracován v rámci projektu EU peníze školám Název školy: Střední zdravotnická škola a Obchodní akademie, Rumburk, příspěvková organizace Registrační číslo projektu: CZ.1.07/1.5.00/34.0649
Výukový materiál zpracován v rámci projektu EU peníze školám Název školy: Střední zdravotnická škola a Obchodní akademie, Rumburk, příspěvková organizace Registrační číslo projektu: CZ.1.07/1.5.00/34.0649
Neočekávaný a nejmenší původce infekčních chorob: prion
 Ze současné medicíny Neočekávaný a nejmenší původce infekčních chorob: prion JAN ŠMARDA Žijeme už v šestnáctém roce jednadvacátého století. A už dvěstě let (díky Louisu Pasteurovi) víme, že původci přenosných
Ze současné medicíny Neočekávaný a nejmenší původce infekčních chorob: prion JAN ŠMARDA Žijeme už v šestnáctém roce jednadvacátého století. A už dvěstě let (díky Louisu Pasteurovi) víme, že původci přenosných
Obecná epidemiologie. MUDr. Miroslava Zavřelová Ústav preventivního lékařství, odd. epidemiologie infekčních chorob
 Obecná epidemiologie MUDr. Miroslava Zavřelová Ústav preventivního lékařství, odd. epidemiologie infekčních chorob Epidemiologie Studium hromadně se vyskytujících jevů Stanovení opatření intervence Analýza
Obecná epidemiologie MUDr. Miroslava Zavřelová Ústav preventivního lékařství, odd. epidemiologie infekčních chorob Epidemiologie Studium hromadně se vyskytujících jevů Stanovení opatření intervence Analýza
NEU/VC hodin praktických cvičení / blok
 Studijní program : Všeobecné lékařství Název předmětu : Neurologie Rozvrhová zkratka : NEU/VC012 Rozvrh výuky : 18 hodin seminářů / blok 72 hodin praktických cvičení / blok Zařazení výuky : 4. ročník,
Studijní program : Všeobecné lékařství Název předmětu : Neurologie Rozvrhová zkratka : NEU/VC012 Rozvrh výuky : 18 hodin seminářů / blok 72 hodin praktických cvičení / blok Zařazení výuky : 4. ročník,
Mgr. Šárka Vopěnková Gymnázium, SOŠ a VOŠ Ledeč nad Sázavou VY_32_INOVACE_02_3_17_BI2 NEMOCI CNS
 Mgr. Šárka Vopěnková Gymnázium, SOŠ a VOŠ Ledeč nad Sázavou VY_32_INOVACE_02_3_17_BI2 NEMOCI CNS ONEMOCNĚNÍ CNS značně různorodé příčiny: vrozené cévní infekční degenerativní poškození CNS má vážné následky
Mgr. Šárka Vopěnková Gymnázium, SOŠ a VOŠ Ledeč nad Sázavou VY_32_INOVACE_02_3_17_BI2 NEMOCI CNS ONEMOCNĚNÍ CNS značně různorodé příčiny: vrozené cévní infekční degenerativní poškození CNS má vážné následky
Péče o pacienty léčené pro demence v ambulantních a lůžkových zařízeních ČR v letech
 Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 31. 12. 2013 57 Souhrn Péče o pacienty léčené pro demence v ambulantních a lůžkových zařízeních ČR v letech 2008 2012
Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 31. 12. 2013 57 Souhrn Péče o pacienty léčené pro demence v ambulantních a lůžkových zařízeních ČR v letech 2008 2012
FUNKČNÍ VARIANTA GENU ANXA11 SNIŽUJE RIZIKO ONEMOCNĚNÍ
 FUNKČNÍ VARIANTA GENU ANXA11 SNIŽUJE RIZIKO ONEMOCNĚNÍ SARKOIDÓZOU: POTVRZENÍ VÝSLEDKŮ CELOGENOMOVÉ ASOCIAČNÍ STUDIE. Sťahelová A. 1, Mrázek F. 1, Kriegová E. 1, Hutyrová B. 2, Kubištová Z. 1, Kolek V.
FUNKČNÍ VARIANTA GENU ANXA11 SNIŽUJE RIZIKO ONEMOCNĚNÍ SARKOIDÓZOU: POTVRZENÍ VÝSLEDKŮ CELOGENOMOVÉ ASOCIAČNÍ STUDIE. Sťahelová A. 1, Mrázek F. 1, Kriegová E. 1, Hutyrová B. 2, Kubištová Z. 1, Kolek V.
Klíšťová encefalitida
 Klíšťová encefalitida Autor: Michaela Měkýšová Výskyt Česká republika patří každoročně mezi státy s vysokým výskytem klíšťové encefalitidy. Za posledních 10 let připadá přibližně 7 nakažených osob na 100
Klíšťová encefalitida Autor: Michaela Měkýšová Výskyt Česká republika patří každoročně mezi státy s vysokým výskytem klíšťové encefalitidy. Za posledních 10 let připadá přibližně 7 nakažených osob na 100
Familiární středomořská (Mediterranean) horečka (Fever)
 horečka (Fever)") www.printo.it/pediatric-rheumatology/cz/intro Familiární středomořská (Mediterranean) horečka (Fever) Verze č 2016 2. DIAGNÓZA A LÉČBA 2.1 Jak se nemoc diagnostikuje? Obecně se uplatňuje následující postup:
www.printo.it/pediatric-rheumatology/cz/intro Familiární středomořská (Mediterranean) horečka (Fever) Verze č 2016 2. DIAGNÓZA A LÉČBA 2.1 Jak se nemoc diagnostikuje? Obecně se uplatňuje následující postup:
Dědičnost vázaná na X chromosom
 12 Dědičnost vázaná na X chromosom EuroGentest - Volně přístupné webové stránky s informacemi o genetickém vyšetření (v angličtině). www.eurogentest.org Orphanet - Volně přístupné webové stránky s informacemi
12 Dědičnost vázaná na X chromosom EuroGentest - Volně přístupné webové stránky s informacemi o genetickém vyšetření (v angličtině). www.eurogentest.org Orphanet - Volně přístupné webové stránky s informacemi
DOPORUČENÝ PRACOVNÍ POSTUP PRO OŠETŘOVÁNÍ PACIENTŮ
 DOPORUČENÝ PRACOVNÍ POSTUP PRO OŠETŘOVÁNÍ PACIENTŮ S PODEZŘENÍM NA CJN, NVCJN A REŽIM DEKONTAMINACE A STERILIZACE MUDR. JAROMÍRA KRATOCHVÍLOVÁ Zpracováno podle: 1. Metodického listu TSE/CJN, Surveillance,
DOPORUČENÝ PRACOVNÍ POSTUP PRO OŠETŘOVÁNÍ PACIENTŮ S PODEZŘENÍM NA CJN, NVCJN A REŽIM DEKONTAMINACE A STERILIZACE MUDR. JAROMÍRA KRATOCHVÍLOVÁ Zpracováno podle: 1. Metodického listu TSE/CJN, Surveillance,
Paliativní péče u nervosvalových onemocnění v dětském věku
 Paliativní péče u nervosvalových onemocnění v dětském věku Jana Haberlová Neuromuskulární centrum FN Motol Klinika dětské neurologie 2.LF UK a FN Motol Obsah přednášky Charakteristika nervosvalových onemocnění
Paliativní péče u nervosvalových onemocnění v dětském věku Jana Haberlová Neuromuskulární centrum FN Motol Klinika dětské neurologie 2.LF UK a FN Motol Obsah přednášky Charakteristika nervosvalových onemocnění
 http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
HIV / AIDS MUDr. Miroslava Zavřelová Ústav preventivního lékařství LF MU
 HIV / AIDS MUDr. Miroslava Zavřelová Ústav preventivního lékařství LF MU e-mail: mizavrel@med.muni.cz I.E.S. Brno, 14. 10. 2014 Historie nákazy 1981 San Francisko, New York mladí pacienti s neobvyklými
HIV / AIDS MUDr. Miroslava Zavřelová Ústav preventivního lékařství LF MU e-mail: mizavrel@med.muni.cz I.E.S. Brno, 14. 10. 2014 Historie nákazy 1981 San Francisko, New York mladí pacienti s neobvyklými
ALZHEIMEROVA CHOROBA. Hana Bibrlová 3.B
 ALZHEIMEROVA CHOROBA Hana Bibrlová 3.B Alzheimerova choroba -neurodegenerativní onemocnění mozku, při kterém dochází k postupné demenci -změny postupně působí rozpad nervových vláken a nervových buněk
ALZHEIMEROVA CHOROBA Hana Bibrlová 3.B Alzheimerova choroba -neurodegenerativní onemocnění mozku, při kterém dochází k postupné demenci -změny postupně působí rozpad nervových vláken a nervových buněk
"Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Základy genetiky, základní pojmy
 "Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Základy genetiky, základní pojmy 1/75 Genetika = věda o dědičnosti Studuje biologickou informaci. Organizmy uchovávají,
"Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Základy genetiky, základní pojmy 1/75 Genetika = věda o dědičnosti Studuje biologickou informaci. Organizmy uchovávají,
Nemoci nervové soustavy. Doc. MUDr. Otakar Keller, CSc.
 Nemoci nervové soustavy Doc. MUDr. Otakar Keller, CSc. MKN 10 - VI.kap.l G00-99 G00-G09 Zánětlivé nemoci centrální nervové soustavy G10-G13 Systémové atrofie postihující primárně nervovou soustavu G20-G26
Nemoci nervové soustavy Doc. MUDr. Otakar Keller, CSc. MKN 10 - VI.kap.l G00-99 G00-G09 Zánětlivé nemoci centrální nervové soustavy G10-G13 Systémové atrofie postihující primárně nervovou soustavu G20-G26
vcjd u anglických dětí
 Studijní materiál důchodce č. 62 Prosinec 2000 vcjd u anglických dětí (Variant Creutzfeldt-Jakob disease in UK children: a national surveillance study) Verity C.M., Nicoll A., Will R.G., Devereux G., Stellitano
Studijní materiál důchodce č. 62 Prosinec 2000 vcjd u anglických dětí (Variant Creutzfeldt-Jakob disease in UK children: a national surveillance study) Verity C.M., Nicoll A., Will R.G., Devereux G., Stellitano
DMPK (ZNF9) V DIFERENCOVANÝCH. Z, Kroupová I, Falk M* M
 V DIFERENCOVANÝCH. Z, Kroupová I, Falk M* M") FISH ANALÝZA m-rna DMPK (ZNF9) V DIFERENCOVANÝCH TKÁNÍCH PACIENT IENTŮ S MYOTONICKOU DYSTROFI FIÍ Lukáš Z, Kroupová I, Falk M* M Ústav patologie FN Brno *Biofyzikáln lní ústav AVČR R Brno Definice MD Myotonická
FISH ANALÝZA m-rna DMPK (ZNF9) V DIFERENCOVANÝCH TKÁNÍCH PACIENT IENTŮ S MYOTONICKOU DYSTROFI FIÍ Lukáš Z, Kroupová I, Falk M* M Ústav patologie FN Brno *Biofyzikáln lní ústav AVČR R Brno Definice MD Myotonická
IMUNOGENETIKA I. Imunologie. nauka o obraných schopnostech organismu. imunitní systém heterogenní populace buněk lymfatické tkáně lymfatické orgány
 IMUNOGENETIKA I Imunologie nauka o obraných schopnostech organismu imunitní systém heterogenní populace buněk lymfatické tkáně lymfatické orgány lymfatická tkáň thymus Imunita reakce organismu proti cizorodým
IMUNOGENETIKA I Imunologie nauka o obraných schopnostech organismu imunitní systém heterogenní populace buněk lymfatické tkáně lymfatické orgány lymfatická tkáň thymus Imunita reakce organismu proti cizorodým
Mutace genu pro Connexin 26 jako významná příčina nedoslýchavosti
 Mutace genu pro Connexin 26 jako významná příčina nedoslýchavosti Petr Lesný 1, Pavel Seeman 2, Daniel Groh 1 1 ORL klinika UK 2. LF a FN Motol Subkatedra dětské ORL IPVZ Přednosta doc. MUDr. Zdeněk Kabelka
Mutace genu pro Connexin 26 jako významná příčina nedoslýchavosti Petr Lesný 1, Pavel Seeman 2, Daniel Groh 1 1 ORL klinika UK 2. LF a FN Motol Subkatedra dětské ORL IPVZ Přednosta doc. MUDr. Zdeněk Kabelka
Současné možnosti léčby a standardní terapie infekce HBV
 Současné možnosti léčby a standardní terapie infekce HBV Prof. MUDr. Petr Husa, CSc. Klinika infekčních chorob LF MU a FN Brno, Kongres adiktologů, Špindlerův mlýn 28.4.2010 Rozdělení virových hepatitid
Současné možnosti léčby a standardní terapie infekce HBV Prof. MUDr. Petr Husa, CSc. Klinika infekčních chorob LF MU a FN Brno, Kongres adiktologů, Špindlerův mlýn 28.4.2010 Rozdělení virových hepatitid
Elektromyografie v diagnostice dědičných neuropatií
 Elektromyografie v diagnostice dědičných neuropatií Mazanec R 1, Seeman P 2, Baránková L 1, Bojar M 1 1 Neurologická klinika 2.LF UK a FN Motol, Praha 2 Klinika dětské neurologie 2.LF UK a FN Motol, Praha
Elektromyografie v diagnostice dědičných neuropatií Mazanec R 1, Seeman P 2, Baránková L 1, Bojar M 1 1 Neurologická klinika 2.LF UK a FN Motol, Praha 2 Klinika dětské neurologie 2.LF UK a FN Motol, Praha
Překrývání neurodegenerativních demencí klinické a neuropatologické aspekty
 Překrývání neurodegenerativních demencí klinické a neuropatologické aspekty Robert Rusina Universita Karlova v Praze, 1. lékařská fakulta a Všeobecná fakultní nemocnice v Praze Neurodegenerace postupný
Překrývání neurodegenerativních demencí klinické a neuropatologické aspekty Robert Rusina Universita Karlova v Praze, 1. lékařská fakulta a Všeobecná fakultní nemocnice v Praze Neurodegenerace postupný
MENDELOVA UNIVERZITA V BRNĚ BAKALÁŘSKÁ PRÁCE
 MENDELOVA UNIVERZITA V BRNĚ BAKALÁŘSKÁ PRÁCE BRNO 2011 LENKA KRUTILOVÁ Mendelova univerzita v Brně Agronomická fakulta Ústav morfologie, fyziologie a genetiky zvířat Detekce polymorfismů prionového genu
MENDELOVA UNIVERZITA V BRNĚ BAKALÁŘSKÁ PRÁCE BRNO 2011 LENKA KRUTILOVÁ Mendelova univerzita v Brně Agronomická fakulta Ústav morfologie, fyziologie a genetiky zvířat Detekce polymorfismů prionového genu
1. Definice a historie oboru molekulární medicína. 3. Základní laboratorní techniky v molekulární medicíně
 Obsah Předmluvy 1. Definice a historie oboru molekulární medicína 1.1. Historie molekulární medicíny 2. Základní principy molekulární biologie 2.1. Historie molekulární biologie 2.2. DNA a chromozomy 2.3.
Obsah Předmluvy 1. Definice a historie oboru molekulární medicína 1.1. Historie molekulární medicíny 2. Základní principy molekulární biologie 2.1. Historie molekulární biologie 2.2. DNA a chromozomy 2.3.
Spinální svalová atrofie. Vypracovali: Kateřina Teplá Monika Madrová Anna Dobrovolná Mária Čižmárová Dominika Štrbová Juraj Štipka
 Spinální svalová atrofie Vypracovali: Kateřina Teplá Monika Madrová Anna Dobrovolná Mária Čižmárová Dominika Štrbová Juraj Štipka Klinický popis - relativně vzácná nemoc, nicméně se jedná o nejčastější
Spinální svalová atrofie Vypracovali: Kateřina Teplá Monika Madrová Anna Dobrovolná Mária Čižmárová Dominika Štrbová Juraj Štipka Klinický popis - relativně vzácná nemoc, nicméně se jedná o nejčastější
Pohled genetika na racionální vyšetřování v preventivní kardiologii
 Pohled genetika na racionální vyšetřování v preventivní kardiologii Tomáš Freiberger Genetická laboratoř, Centrum kardiovaskulární a transplantační chirurgie Brno, ČR Osnova Genetické faktory vzniku KV
Pohled genetika na racionální vyšetřování v preventivní kardiologii Tomáš Freiberger Genetická laboratoř, Centrum kardiovaskulární a transplantační chirurgie Brno, ČR Osnova Genetické faktory vzniku KV
MUDr. Helena Šutová Laboratoře Mikrochem a.s.
 Laboratorní diagnostika celiakie MUDr. Helena Šutová Laboratoře Mikrochem a.s. Celiakie Autoimunní onemocnění způsobené požitím lepku (glutenu) s typickým zánětlivým postižením tenkého střeva s genetickou
Laboratorní diagnostika celiakie MUDr. Helena Šutová Laboratoře Mikrochem a.s. Celiakie Autoimunní onemocnění způsobené požitím lepku (glutenu) s typickým zánětlivým postižením tenkého střeva s genetickou
Varovné signály (Red flags) pro klinickou praxi vodítko pro zvýšené riziko genetické příčiny onemocnění u pacienta
 pro klinickou praxi vodítko pro zvýšené riziko genetické příčiny onemocnění u pacienta") Varovné signály (Red flags) pro klinickou praxi vodítko pro zvýšené riziko genetické příčiny onemocnění u pacienta Obecné varovné signály pro klinickou praxi Přítomnost jednoho nebo více varovných signálů
Varovné signály (Red flags) pro klinickou praxi vodítko pro zvýšené riziko genetické příčiny onemocnění u pacienta Obecné varovné signály pro klinickou praxi Přítomnost jednoho nebo více varovných signálů
Deficit antagonisty IL-1 receptoru (DIRA)
") www.printo.it/pediatric-rheumatology/cz/intro Deficit antagonisty IL-1 receptoru (DIRA) Verze č 2016 1. CO JE DIRA? 1.1 O co se jedná? Deficit antagonisty IL-1Receptoru (DIRA) je vzácné vrozené onemocnění.
www.printo.it/pediatric-rheumatology/cz/intro Deficit antagonisty IL-1 receptoru (DIRA) Verze č 2016 1. CO JE DIRA? 1.1 O co se jedná? Deficit antagonisty IL-1Receptoru (DIRA) je vzácné vrozené onemocnění.
Výukový materiál zpracován v rámci projektu EU peníze školám
 http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
http://vtm.zive.cz/aktuality/vzorek-dna-prozradi-priblizny-vek-pachatele Autorem materiálu a všech jeho částí, není-li uvedeno jinak, je Mgr. Eva Strnadová. Dostupné z Metodického portálu www.rvp.cz ;
AUG STOP AAAA S S. eukaryontní gen v genomové DNA. promotor exon 1 exon 2 exon 3 exon 4. kódující oblast. introny
 eukaryontní gen v genomové DNA promotor exon 1 exon 2 exon 3 exon 4 kódující oblast introny primární transkript (hnrna, pre-mrna) postranskripční úpravy (vznik maturované mrna) syntéza čepičky AUG vyštěpení
eukaryontní gen v genomové DNA promotor exon 1 exon 2 exon 3 exon 4 kódující oblast introny primární transkript (hnrna, pre-mrna) postranskripční úpravy (vznik maturované mrna) syntéza čepičky AUG vyštěpení
Návrh směrnic pro správnou laboratorní diagnostiku Friedreichovy ataxie.
 Návrh směrnic pro správnou laboratorní diagnostiku Friedreichovy ataxie. Připravila L.Fajkusová Online Mendelian Inheritance in Man: #229300 FRIEDREICH ATAXIA 1; FRDA *606829 FRDA GENE; FRDA Popis onemocnění
Návrh směrnic pro správnou laboratorní diagnostiku Friedreichovy ataxie. Připravila L.Fajkusová Online Mendelian Inheritance in Man: #229300 FRIEDREICH ATAXIA 1; FRDA *606829 FRDA GENE; FRDA Popis onemocnění
Péče o pacienty s diagnózami F01, F03 a G30 (demence) v lůžkových zařízeních ČR v letech
 v lůžkových zařízeních ČR v letech") Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 14. 12. 2011 61 Péče o pacienty s diagnózami, a (demence) v lůžkových zařízeních ČR v letech 2006 2010 Health care
Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 14. 12. 2011 61 Péče o pacienty s diagnózami, a (demence) v lůžkových zařízeních ČR v letech 2006 2010 Health care
Hereditární spastická paraparéza. Stanislav Voháňka
 Hereditární spastická paraparéza Stanislav Voháňka HSPP (hereditární spastická paraplegie) Skupina dědičných chorob Progredující spastická paraparéza Synonyma Familiární spastická paraplegie Onemocnění
Hereditární spastická paraparéza Stanislav Voháňka HSPP (hereditární spastická paraplegie) Skupina dědičných chorob Progredující spastická paraparéza Synonyma Familiární spastická paraplegie Onemocnění
Masivně paralelní sekvenování v diagnostice závažných časných epilepsií. DNA laboratoř KDN 2.LF a FN v Motole
 Masivně paralelní sekvenování v diagnostice závažných časných epilepsií DNA laboratoř KDN 2.LF a FN v Motole Financial disclosure (konflikt zájmů) Projekt je v současné době finančně zabezpečen pouze z
Masivně paralelní sekvenování v diagnostice závažných časných epilepsií DNA laboratoř KDN 2.LF a FN v Motole Financial disclosure (konflikt zájmů) Projekt je v současné době finančně zabezpečen pouze z
po přisátí klíštěte virus prokazován kůže virémie buňky RES (slezina, játra, kostní dřeň) mechanismus invaze do CNS nejasný, HEB při virémii
 mechanismus invaze do CNS nejasný, HEB při virémii") Středoevropská klíšťová meningoencefalitida klinický obraz MUDr. Dita Smíšková, PhD Doc.MUDr.Vilma Marešová, CSc. MEK patogeneze po přisátí klíštěte virus prokazován nejprve v Langerhansových h buňkách
Středoevropská klíšťová meningoencefalitida klinický obraz MUDr. Dita Smíšková, PhD Doc.MUDr.Vilma Marešová, CSc. MEK patogeneze po přisátí klíštěte virus prokazován nejprve v Langerhansových h buňkách
10. přehledu o provedení krevní transfúze v uplynulých
 Strana 1236 Sbírka zákonů č. 114 / 2013 Částka 51 114 VYHLÁŠKA ze dne 29. dubna 2013 o stanovení bližších podmínek posuzování zdravotní způsobilosti a rozsahu vyšetření žijícího nebo zemřelého dárce tkání
Strana 1236 Sbírka zákonů č. 114 / 2013 Částka 51 114 VYHLÁŠKA ze dne 29. dubna 2013 o stanovení bližších podmínek posuzování zdravotní způsobilosti a rozsahu vyšetření žijícího nebo zemřelého dárce tkání
GAUCHEROVA CHOROBA A VAŠE RODINA
 GAUCHEROVA CHOROBA VČASNÁ DIAGNÓZA Připravila a financovala společnost Shire. GAUCHEROVA CHOROBA A VAŠE RODINA Informační brožura pro pacienty s Gaucherovou chorobou Gaucherova choroba je vzácné dědičné
GAUCHEROVA CHOROBA VČASNÁ DIAGNÓZA Připravila a financovala společnost Shire. GAUCHEROVA CHOROBA A VAŠE RODINA Informační brožura pro pacienty s Gaucherovou chorobou Gaucherova choroba je vzácné dědičné
PAS v každodenní praxi dětské psychiatrie EVA ČÁPOVÁ
 PAS v každodenní praxi dětské psychiatrie EVA ČÁPOVÁ Poruchy autistického spektra Všepronikající hrubá neurovývojová porucha mozku PAS (autistic spektrum disorder ASD) 1979 Lorna Wing a Judith Gould Výskyt
PAS v každodenní praxi dětské psychiatrie EVA ČÁPOVÁ Poruchy autistického spektra Všepronikající hrubá neurovývojová porucha mozku PAS (autistic spektrum disorder ASD) 1979 Lorna Wing a Judith Gould Výskyt
CÉVNÍ MALFORMACE MOZKU - KAVERNOMY
 CÉVNÍ MALFORMACE MOZKU - KAVERNOMY E.Vítková, D.Krajíčková, J.Náhlovský Neurologická a Neurochirurgická klinika LF UK a FN Hradec Králové Kavernomy Makroskopicky Morušovitý útvar mm až několik cm Dutinky
CÉVNÍ MALFORMACE MOZKU - KAVERNOMY E.Vítková, D.Krajíčková, J.Náhlovský Neurologická a Neurochirurgická klinika LF UK a FN Hradec Králové Kavernomy Makroskopicky Morušovitý útvar mm až několik cm Dutinky
Obr.1 Žilní splavy. https://s-media-cache-ak0.pinimg.com/564x/c3/91/8c/c3918c00db875bb460cf868b26ee1a0c.jpg
 TROMBÓZA NITROLEBNÍCH ŽIL A SPLAVŮ Autor: Barbora Baštinská Výskyt Mozková žilní trombóza je vzácné onemocnění, jehož příznaky se mohou značně lišit. Vyskytuje se spíše u mladších pacientů a většinou (až
TROMBÓZA NITROLEBNÍCH ŽIL A SPLAVŮ Autor: Barbora Baštinská Výskyt Mozková žilní trombóza je vzácné onemocnění, jehož příznaky se mohou značně lišit. Vyskytuje se spíše u mladších pacientů a většinou (až
GENETIKA. Dědičnost a pohlaví
 GENETIKA Dědičnost a pohlaví Chromozómové určení pohlaví Dvoudomé rostliny a gonochoristé (živočichové odděleného pohlaví) mají pohlaví určeno dědičně chromozómovou výbavou jedince = dvojicí pohlavních
GENETIKA Dědičnost a pohlaví Chromozómové určení pohlaví Dvoudomé rostliny a gonochoristé (živočichové odděleného pohlaví) mají pohlaví určeno dědičně chromozómovou výbavou jedince = dvojicí pohlavních
Péče o pacienty s diagnózami F01, F03 a G30 - demence v lůžkových zařízeních ČR v letech
 Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 20. 12. 2010 68 Péče o pacienty s diagnózami, a - demence v lůžkových zařízeních ČR v letech 2005 2009 Health care
Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 20. 12. 2010 68 Péče o pacienty s diagnózami, a - demence v lůžkových zařízeních ČR v letech 2005 2009 Health care
Strašák EBOLA TÝKÁ SE TAKÉ NÁS EVROPANY? Bc. Helena Marcinková
 Strašák EBOLA TÝKÁ SE TAKÉ NÁS EVROPANY? Bc. Helena Marcinková Ebola a Česká republika máme se bát? Jaké je riziko, že se Ebola dostane do České republiky a začne se tu šířit? Riziko pro turisty nebo obchodní
Strašák EBOLA TÝKÁ SE TAKÉ NÁS EVROPANY? Bc. Helena Marcinková Ebola a Česká republika máme se bát? Jaké je riziko, že se Ebola dostane do České republiky a začne se tu šířit? Riziko pro turisty nebo obchodní
Kazuistiky sporadické varianty Creutzfeldtovy-Jakobovy nemoci
 292 Kazuistiky sporadické varianty Creutzfeldtovy-Jakobovy nemoci MUDr. Tereza Lukoszová Neurologické oddělení, Nemocnice Havlíčkův Brod Prionová onemocnění představují skupinu vzácných neurodegenerativních
292 Kazuistiky sporadické varianty Creutzfeldtovy-Jakobovy nemoci MUDr. Tereza Lukoszová Neurologické oddělení, Nemocnice Havlíčkův Brod Prionová onemocnění představují skupinu vzácných neurodegenerativních
Úvod do nonhla-dq genetiky celiakie
 Úvod do nonhla-dq genetiky celiakie František Mrázek HLA laboratoř, Ústav Imunologie LF UP a FN Olomouc Celiakie - časté chronické zánětlivé onemocnění tenkého střeva s autoimunitní a systémovou složkou
Úvod do nonhla-dq genetiky celiakie František Mrázek HLA laboratoř, Ústav Imunologie LF UP a FN Olomouc Celiakie - časté chronické zánětlivé onemocnění tenkého střeva s autoimunitní a systémovou složkou
Kazuistiky sporadické varianty Creutzfeldtovy-Jakobovy nemoci
 Kazuistiky sporadické varianty Creutzfeldtovy-Jakobovy nemoci MUDr. Tereza Lukoszová Neurologické oddělení, Nemocnice Havlíčkův Brod Prionová onemocnění představují skupinu vzácných neurodegenerativních
Kazuistiky sporadické varianty Creutzfeldtovy-Jakobovy nemoci MUDr. Tereza Lukoszová Neurologické oddělení, Nemocnice Havlíčkův Brod Prionová onemocnění představují skupinu vzácných neurodegenerativních
Výskyt a význam infekce Borna disease virem u pacientů léčených
 Výskyt a význam infekce Borna disease virem u pacientů léčených pro závislost Sylva Racková Psychiatrická klinika LF UK v Plzni AT konference 28.04. 2010, Špindlerův Mlýn Borna Disease virus (BDV) charakteristika
Výskyt a význam infekce Borna disease virem u pacientů léčených pro závislost Sylva Racková Psychiatrická klinika LF UK v Plzni AT konference 28.04. 2010, Špindlerův Mlýn Borna Disease virus (BDV) charakteristika
Deoxyribonukleová kyselina (DNA)
") Genetika Dědičností rozumíme schopnost rodičů předávat své vlastnosti potomkům a zachovat tak rozličnost druhů v přírodě. Dědičností a proměnlivostí jedinců se zabývá vědní obor genetika. Základní jednotkou
Genetika Dědičností rozumíme schopnost rodičů předávat své vlastnosti potomkům a zachovat tak rozličnost druhů v přírodě. Dědičností a proměnlivostí jedinců se zabývá vědní obor genetika. Základní jednotkou
Převzato na základě svolení Macmillan Publishers Ltd: Nat Rev Genet. 2001;2(4):245-55), copyright (2001).
:245-55), copyright (2001).") α-talasemie Autor: Alexandra Kredátusová Výskyt Talasemie jsou světově nejrozšířenější dědičně podmíněné krevní onemocnění. Nejčastější jsou v malarických oblastech (obr. 1), protože nositeli talasemického
α-talasemie Autor: Alexandra Kredátusová Výskyt Talasemie jsou světově nejrozšířenější dědičně podmíněné krevní onemocnění. Nejčastější jsou v malarických oblastech (obr. 1), protože nositeli talasemického
STŘEDNÍ ZDRAVOTNICKÁ ŠKOLA A VYŠŠÍ ODBORNÁ ŠKOLA ZDRAVOTNICKÁ ŽĎÁR NAD SÁZAVOU OBECNÁ EPIDEMIOLOGIE MGR. IVA COUFALOVÁ
 STŘEDNÍ ZDRAVOTNICKÁ ŠKOLA A VYŠŠÍ ODBORNÁ ŠKOLA ZDRAVOTNICKÁ ŽĎÁR NAD SÁZAVOU OBECNÁ EPIDEMIOLOGIE MGR. IVA COUFALOVÁ EPIDEMIOLOGIE je obor, který zkoumá rozložení infekčních chorob v populaci, sleduje
STŘEDNÍ ZDRAVOTNICKÁ ŠKOLA A VYŠŠÍ ODBORNÁ ŠKOLA ZDRAVOTNICKÁ ŽĎÁR NAD SÁZAVOU OBECNÁ EPIDEMIOLOGIE MGR. IVA COUFALOVÁ EPIDEMIOLOGIE je obor, který zkoumá rozložení infekčních chorob v populaci, sleduje
Biologie - Oktáva, 4. ročník (humanitní větev)
") - Oktáva, 4. ročník (humanitní větev) Biologie Výchovné a vzdělávací strategie Kompetence k řešení problémů Kompetence komunikativní Kompetence sociální a personální Kompetence občanská Kompetence k podnikavosti
- Oktáva, 4. ročník (humanitní větev) Biologie Výchovné a vzdělávací strategie Kompetence k řešení problémů Kompetence komunikativní Kompetence sociální a personální Kompetence občanská Kompetence k podnikavosti
Predikce epidemií listerióz molekulárně biologickými metodami
 Predikce epidemií listerióz molekulárně biologickými metodami Renáta Karpíšková Tereza Gelbíčová Molekulárně-biologické metody v surveillance a šetření epidemií, SZÚ Praha, 28. 3. 2019 Listerióza invazivní
Predikce epidemií listerióz molekulárně biologickými metodami Renáta Karpíšková Tereza Gelbíčová Molekulárně-biologické metody v surveillance a šetření epidemií, SZÚ Praha, 28. 3. 2019 Listerióza invazivní
Merosin deficitní kongenitální svalová dystrofie.
 Merosin deficitní kongenitální svalová dystrofie www.adamhrdy.cz Narození a určení diagnozy Adámek se narodil jako prvorozený syn, dne 19.8.2013 v Thomayerově nemocnici v Krči. Po narození nic nenasvědčovalo
Merosin deficitní kongenitální svalová dystrofie www.adamhrdy.cz Narození a určení diagnozy Adámek se narodil jako prvorozený syn, dne 19.8.2013 v Thomayerově nemocnici v Krči. Po narození nic nenasvědčovalo
Novinky v klasifikaci NSCLC, multidisciplinární konsenzus. testování NSCLC
 Novinky v klasifikaci NSCLC, multidisciplinární konsenzus 2012 pro molekulární testování NSCLC Radoslav Matěj Oddělení patologie a mol. medicíny Thomayerovy nemocnice, Praha Konsenzus Čestlice 25.6. 2010
Novinky v klasifikaci NSCLC, multidisciplinární konsenzus 2012 pro molekulární testování NSCLC Radoslav Matěj Oddělení patologie a mol. medicíny Thomayerovy nemocnice, Praha Konsenzus Čestlice 25.6. 2010
Obr. 1 Vzorec adrenalinu
 Feochromocytom, nádor nadledvin Autor: Antonín Zdráhal Výskyt Obecně nádorové onemocnění vzniká následkem nekontrolovatelného množení buněk, k němuž dochází mnoha různými mechanismy, někdy tyto příčiny
Feochromocytom, nádor nadledvin Autor: Antonín Zdráhal Výskyt Obecně nádorové onemocnění vzniká následkem nekontrolovatelného množení buněk, k němuž dochází mnoha různými mechanismy, někdy tyto příčiny
Využití DNA markerů ve studiu fylogeneze rostlin
 Mendelova genetika v příkladech Využití DNA markerů ve studiu fylogeneze rostlin Ing. Petra VESELÁ Ústav lesnické botaniky, dendrologie a geobiocenologie LDF MENDELU Brno Tento projekt je spolufinancován
Mendelova genetika v příkladech Využití DNA markerů ve studiu fylogeneze rostlin Ing. Petra VESELÁ Ústav lesnické botaniky, dendrologie a geobiocenologie LDF MENDELU Brno Tento projekt je spolufinancován
MUDr. Milena Bretšnajdrová, Ph.D. Prim. MUDr. Zdeněk Záboj. Odd. geriatrie Fakultní nemocnice Olomouc
 MUDr. Milena Bretšnajdrová, Ph.D. Prim. MUDr. Zdeněk Záboj Odd. geriatrie Fakultní nemocnice Olomouc Neurodegenerativní onemocnění mozku, při kterém dochází k postupné demenci. V patofyziologickém obraze
MUDr. Milena Bretšnajdrová, Ph.D. Prim. MUDr. Zdeněk Záboj Odd. geriatrie Fakultní nemocnice Olomouc Neurodegenerativní onemocnění mozku, při kterém dochází k postupné demenci. V patofyziologickém obraze
Papillomaviry. Eva Hamšíková. ÚHKT, oddělení experimentální virologie
 Papillomaviry Eva Hamšíková ÚHKT, oddělení experimentální virologie Viry nitrobuněční parazité, životní cyklus závislý na buněčném aparátu, v současnosti známo asi 5 000 typů virů Papillomaviry malé viry
Papillomaviry Eva Hamšíková ÚHKT, oddělení experimentální virologie Viry nitrobuněční parazité, životní cyklus závislý na buněčném aparátu, v současnosti známo asi 5 000 typů virů Papillomaviry malé viry
Periodické syndromy asociované s kryopyrinem (CAPS)
") www.printo.it/pediatric-rheumatology/cz/intro Periodické syndromy asociované s kryopyrinem (CAPS) Verze č 2016 1. CO JE CAPS 1.1 Co je to? Periodické syndromy asociované s kryopyrinem (CAPS) nebo také
www.printo.it/pediatric-rheumatology/cz/intro Periodické syndromy asociované s kryopyrinem (CAPS) Verze č 2016 1. CO JE CAPS 1.1 Co je to? Periodické syndromy asociované s kryopyrinem (CAPS) nebo také
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/
 I n v e s t i c e d o r o z v o j e v z d ě l á v á n í Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním
I n v e s t i c e d o r o z v o j e v z d ě l á v á n í Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním
ÚVOD DO TRANSPLANTAČNÍ IMUNOLOGIE
 ÚVOD DO TRANSPLANTAČNÍ IMUNOLOGIE Základní funkce imunitního systému Chrání integritu organizmu proti škodlivinám zevního a vnitřního původu: chrání organizmus proti patogenním mikroorganizmům a jejich
ÚVOD DO TRANSPLANTAČNÍ IMUNOLOGIE Základní funkce imunitního systému Chrání integritu organizmu proti škodlivinám zevního a vnitřního původu: chrání organizmus proti patogenním mikroorganizmům a jejich
Health care about patients with eating disorders in the Czech Republic in
 Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 11. 10. 2017 6 Péče o pacienty s poruchami příjmu potravy v ČR v letech 2010 2016 Health care about patients with eating
Aktuální informace Ústavu zdravotnických informací a statistiky České republiky Praha 11. 10. 2017 6 Péče o pacienty s poruchami příjmu potravy v ČR v letech 2010 2016 Health care about patients with eating
Beličková 1, J Veselá 1, E Stará 1, Z Zemanová 2, A Jonášová 2, J Čermák 1
 Beličková 1, J Veselá 1, E Stará 1, Z Zemanová 2, A Jonášová 2, J Čermák 1 1 Ústav hematologie a krevní transfuze, Praha 2 Všeobecná fakultní nemocnice, Praha MDS Myelodysplastický syndrom (MDS) je heterogenní
Beličková 1, J Veselá 1, E Stará 1, Z Zemanová 2, A Jonášová 2, J Čermák 1 1 Ústav hematologie a krevní transfuze, Praha 2 Všeobecná fakultní nemocnice, Praha MDS Myelodysplastický syndrom (MDS) je heterogenní
Martina Havlíčková Helena Jiřincová. NRL pro chřipku, Státní zdravotní ústav
 Pandemic H1N1 2009 Martina Havlíčková Helena Jiřincová NRL pro chřipku, Státní zdravotní ústav Z historie H1N1 1916-1917 pravděpodobná cirkulace viru, malá ohniska, lokální epidemie ve vojenských táborech,
Pandemic H1N1 2009 Martina Havlíčková Helena Jiřincová NRL pro chřipku, Státní zdravotní ústav Z historie H1N1 1916-1917 pravděpodobná cirkulace viru, malá ohniska, lokální epidemie ve vojenských táborech,
Patient s hemato-onkologickým onemocněním: péče v závěru života - umírání v ČR, hospicová péče - zkušenosti jednoho pracoviště
 Patient s hemato-onkologickým onemocněním: péče v závěru života - umírání v ČR, hospicová péče - zkušenosti jednoho pracoviště Jindřich Polívka 1, Jan Švancara 2 1 Hospic Dobrého Pastýře Čerčany 2 Institut
Patient s hemato-onkologickým onemocněním: péče v závěru života - umírání v ČR, hospicová péče - zkušenosti jednoho pracoviště Jindřich Polívka 1, Jan Švancara 2 1 Hospic Dobrého Pastýře Čerčany 2 Institut
 www.printo.it/pediatric-rheumatology/cz/intro Majeedův Verze č 2016 1. CO JE MAJEEDŮV SYNDROM? 1.1 Co je to? Majeedův syndrom je vzácné, geneticky podmíněné onemocnění. Pacienti mají projevy chronické
www.printo.it/pediatric-rheumatology/cz/intro Majeedův Verze č 2016 1. CO JE MAJEEDŮV SYNDROM? 1.1 Co je to? Majeedův syndrom je vzácné, geneticky podmíněné onemocnění. Pacienti mají projevy chronické
Diferenciální diagnostika parkinsonských syndromů. Jiří Klempíř
 Diferenciální diagnostika parkinsonských syndromů Jiří Klempíř Parkinsonský syndrom Primární Sekundární Parkinsonova nemoc Parkinsonský syndrom Sporadická? Hereditární sporadický hereditární MSA Wilsonova
Diferenciální diagnostika parkinsonských syndromů Jiří Klempíř Parkinsonský syndrom Primární Sekundární Parkinsonova nemoc Parkinsonský syndrom Sporadická? Hereditární sporadický hereditární MSA Wilsonova
Genetika kardiomyopatií. Pavol Tomašov Kardiologická klinika 2. LF UK a FN v Motole, Praha
 Genetika kardiomyopatií Pavol Tomašov Kardiologická klinika 2. LF UK a FN v Motole, Praha Úvod 1. Některé kardiomyopatie jsou monogenně podmíněná dědičná onemocnění 2. Dědičné kardiomyopatie mají velkou
Genetika kardiomyopatií Pavol Tomašov Kardiologická klinika 2. LF UK a FN v Motole, Praha Úvod 1. Některé kardiomyopatie jsou monogenně podmíněná dědičná onemocnění 2. Dědičné kardiomyopatie mají velkou
Cvičeníč. 10 Dědičnost a pohlaví. Mgr. Zbyněk Houdek
 Cvičeníč. 10 Dědičnost a pohlaví Mgr. Zbyněk Houdek Dědičnost a pohlaví Gonozomy se v evoluci vytvořily z autozomů, proto obsahují nejen geny řídící vznik pohlavních rozdílů, ale i další geny. V těchto
Cvičeníč. 10 Dědičnost a pohlaví Mgr. Zbyněk Houdek Dědičnost a pohlaví Gonozomy se v evoluci vytvořily z autozomů, proto obsahují nejen geny řídící vznik pohlavních rozdílů, ale i další geny. V těchto
PŘEDBĚŽNÝ NÁVRH USNESENÍ
 Evropský parlament 2014-2019 Výbor pro životní prostředí, veřejné zdraví a bezpečnost potravin 25.5.2018 2018/0000(RSP) PŘEDBĚŽNÝ NÁVRH USNESENÍ předložený na základě otázky k ústnímu zodpovězení B8-0000/2018
Evropský parlament 2014-2019 Výbor pro životní prostředí, veřejné zdraví a bezpečnost potravin 25.5.2018 2018/0000(RSP) PŘEDBĚŽNÝ NÁVRH USNESENÍ předložený na základě otázky k ústnímu zodpovězení B8-0000/2018
Význam genetického vyšetření u pacientů s mentální retardací
 Význam genetického vyšetření u pacientů s mentální retardací Šantavá, A., Hyjánek, J., Čapková, P., Adamová, K., Vrtěl, R. Ústav lékařské genetiky a fetální medicíny FN a LF UP Olomouc Mentální retardace
Význam genetického vyšetření u pacientů s mentální retardací Šantavá, A., Hyjánek, J., Čapková, P., Adamová, K., Vrtěl, R. Ústav lékařské genetiky a fetální medicíny FN a LF UP Olomouc Mentální retardace
Lymeská borrelióza epidemiologická data za rok 2014
 Lymeská borrelióza epidemiologická data za rok 2014 Mapy a grafy incidencí lymeské borreliózy v České republice. Maps and graphs of lyme borreliosis (Lyme disease) in the Czech Republic. 15.září 2015 Doc.MUDr.Bohumír
Lymeská borrelióza epidemiologická data za rok 2014 Mapy a grafy incidencí lymeské borreliózy v České republice. Maps and graphs of lyme borreliosis (Lyme disease) in the Czech Republic. 15.září 2015 Doc.MUDr.Bohumír