UNIVERZITA KARLOVA V PRAZE FARMACEUTICKÁ FAKULTA V HRADCI KRÁLOVÉ
|
|
|
- Vít Mašek
- před 6 lety
- Počet zobrazení:
Transkript
1 UNIVERZITA KARLOVA V PRAZE FARMACEUTICKÁ FAKULTA V HRADCI KRÁLOVÉ Katedra analytické chemie Optimalizace kroku přípravy vzorku biologického materiálu pro UHPLC-MS/MS analýzu atorvastatinu, rosuvastatinu a jejich metabolitů Vedoucí diplomové práce: Doc. PharmDr. Lucie Nováková Ph.D. Vedoucí katedry: Prof. RNDr. Petr Solich CSc. Hradec Králové Veronika Pilařová
2 ABSTRAKT Univerzita Karlova v Praze, Farmaceutická fakulta v Hradci Králové Katedra analytické chemie Kandidát: Veronika Pilařová Školitel: Doc. PharmDr. Lucie Nováková Ph.D. Název diplomové práce: Optimalizace kroku přípravy vzorku biologického materiálu pro UHPLC-MS/MS analýzu atorvastatinu, rosuvastatinu a jejich metabolitů. Cílem této diplomové práce bylo optimalizovat UHPLC-MS/MS metodu určenou pro stanovení koncentrace atorvastatinu, rosuvastatinu a jejich metabolitů v biologickém materiálu a dále vyvinout a optimalizovat metodu pro přípravu vzorku biologického materiálu technikou MEPS, t. j. mikroextrakcí pomocí tuhého sorbentu, a tuto metodu zvalidovat. K separaci analytů byla využita kolona Acquity BEH C18 (50 x 2,1 mm, 1,7 µm, Waters). Ionizace analytů byla provedena elektrosprejem v pozitivním i v negativním módu. Jako detektor byl použit trojitý kvadrupól. U každého analytu byl zvolen prekurzorový ion, ion fragmentu a optimalizována kolizní energie a napětí na vstupním kuželu pro každou látku zvlášť. Kvantifikace látek byla provedena pomocí SRM (monitorování vybrané reakce). Pro rosuvastatin lakton, který byl stanovován v pozitivním módu, byla prekurzorovým iontem zvolena protonovaná molekula [M+H] +. Pro zbylé statiny byl v negativním módu vybrán prekurzorový ion [M-H] -, který měl největší intenzitu v hmotnostním spektru. Tyto podmínky byly využity pro vývoj a optimalizaci metody MEPS. Byly testovány tuhé fáze C18, C8 a M1. Nejlepší výsledky vykazoval sorbent C8. Byla zkoušena promývací a eluční činidla složená z acetonitrilu a octanu amonného v různých poměrech. Jako eluční činidlo byl zvolen roztok acetonitrilu a 0,1M octanu amonného o ph 4,5 v poměru 95:5. Jako promývací činidlo byl vybrán roztok acetonitrilu a 0,01M octanu amonného o ph 4,5 v poměru 5:95. Po optimalizaci těchto kroků byla metoda aplikována na biologický materiál, v tomto případě na sérum. Metoda byla zvalidována. Byla ověřena linearita, správnost, přesnost a selektivita metody. Byl stanoven limit detekce a kvantifikace analytů a matricové efekty. Metoda byla lineární. Korelační koeficienty byly stanoveny v rozmezí 0,9982 0,9998. Hodnoty detekčního limitu (LOD) byly v rozmezí 0,15 0,75 nmol/l a hodnoty kvantitativního limitu (LOQ) 0,5-2,5 nmol/l. Klíčová slova: MEPS, UHPLC-MS/MS, rosuvastatin, rosuvastatin lakton, N-desmethyl rosuvastatin, atorvastatin, atorvastatin lakton, p-hydroxyatorvastatin, o-hydroxyatorvastatin
3 ABSTRACT Charles University in Prague, Faculty of Pharmacy in Hradec Králové Department of analytical chemistry Candidate: Veronika Pilařová Supervisor: Doc. PharmDr. Lucie Nováková Ph.D. Title of GraduationThesis: Optimalization of sample preparation step for UHPLC-MS/MS analysis of atorvastatin, rosuvastatin and their metabolites The purpose of this graduation thesis was the optimalization of UHPLC-MS/MS method for the determination of concentrations of atorvastatin, rosuvastatin and their metabolites in biological material, and then development and optimalization of a MEPS (microextraction by packed sorbent) method for sample preparation of biological material and validation of this method. Acquity BEH C18 column (50 x 2.1 mm, 1.7 µm, Waters) was used for the separation of the analytes. Electrospray ionization was performed in both negative and positive ion mode. Triple quadrupole mass analyser was used for detection. Precursor ions and fragment ions were chosen for each statin. Collision energy and cone voltage were optimized for all analytes individually. Quantification of analytes was performed using the SRM (selected reaction monitoring). Protonated molecule [M+H] +, which was measured in the positive ion mode, was chosen as a precursor ion for rosuvastatin lactone. Ion [M-H] -, which was measured in the negative mode, was selected for the other statins, because it was the most intense ion in the mass spectrum. These conditions were used for the development and optimalization of MEPS method. Following solid phases were tested - C18, C8 and M1. C8 sorbent provided the best results. Washing and elution reagents consisting of acetonitrile and ammonium acetate in various ratios were tested. The mixture of acetonitrile and 0.1 M ammonium acetate ph 4.5 in a ratio of 95:5 was chosen as elution agent. A mixture of acetonitrile and 0.01M ammonium acetate ph 4.5 in ratio of 5:95 was chosen as washing agent. The method was applied to the biological material after optimalization. In this case it was a human serum. The method was validated. The linearity, accuracy, precision and selectivity of the method were verified. The limit of detection and quantification and matrix effects were verified too. The method was linear. Correlation coefficients were determined in the range from to Limit of detection (LOD) ranged from 0.15 to 0.75 nmol / l and limit of quantification (LOQ) from 0.5 to 2.5 nmol / l.
4 Key words: MEPS, UHPLC-MS/MS, rosuvastatin, rosuvastatin lactone, N-desmethyl rosuvastatin, atorvastatin, atorvastatin lactone, p-hydroxyatorvastatin, o- hydroxyatorvastatin
5 Prohlašuji, že tato práce je mým původním autorským dílem. Veškerá literatura a další zdroje, z nichž jsem při zpracování čerpala, jsou uvedeny v seznamu použité literatury a v práci řádně citovány. Práce nebyla využita k získání jiného nebo stejného titulu. Ve Vejvanovicích dne Veronika Pilařová
6 Děkuji Doc. PharmDr. Lucii Novákové Ph.D. za odborné vedení, poskytnutí pomoci a rad, za spolupráci, vstřícné jednání a čas, který mi věnovala během vypracování mojí diplomové práce. Také děkuji RNDr. Haně Vlčkové Ph.D. za cenné rady, spolupráci a příjemnou pracovní atmosféru. Velké poděkování patří také mé rodině, která při mně stála a při studiu mě podporovala.
7 OBSAH Seznam zkratek Úvod Cíl a zadání práce Teoretická část Statiny Struktura statinů Fyzikálně chemické vlastnosti analyzovaných statinů Mechanismus účinku statinů Farmakologické účinky a klinické využití statinů Metody stanovení rosuvastatinu a atorvastatinu Ultra vysokoúčinná kapalinová chromatografie (UHPLC) Hmotnostní spektrometrie Instrumentace MS Spojení kapalinové chromatografie a hmotnostní spektrometrie Způsoby úpravy vzorků Konvenční techniky Moderní techniky Validace metody a matricové efekty Validace analytické metody Matricové efekty Experimentální část Materiál a pomůcky Chemikálie Přístroje a pomůcky Příprava roztoků Příprava zásobních roztoků Příprava pracovních roztoků Příprava mobilní fáze Optimalizace UHPLC-MS podmínek Optimalizace metody MEPS Opakovatelnost metody v rámci SST Validace metody Výsledky a diskuze Výchozí podmínky metody UHPLC-MS/MS UHPLC separace... 48
8 5.1.2 Tandemová hmotnostní detekce Použití vnitřních standardů Optimalizace MEPS Výběr sorbentu a optimalizace elučního činidla Optimalizace promývacího činidla a konečná volba sorbentu Ověření na biologickém materiálu Opakovatelnost a validace metody Opakovatelnost metody Validace metody Závěr Seznam použité literatury Přílohy Abstrakty a publikace... 72
9 Seznam zkratek ACN AmAc APCI APPI AT AT-d 5 ATL BEH ČL DAD DES RV DI-SDME DLLME DPX EČ EI EMA ESI FAc HDL HF-LLLME HF-LPME HILIC Acetonitril Octan amonný Chemická ionizace za atmosférického tlaku (Atmospheric pressure chemical ionization) Fotoionizace za atmosférického tlaku (Atmospheric pressure photoionization) Atorvastatin Deuteriem značený atorvastatin Atorvastatin lakton Hybridní stacionární fáze (Bridged ethyl hybrid) Český lékopis Detektor diodového pole (Diode array detector) N-desmethylrosuvastatin SDME provedené pomocí přímého ponoření (Direct immersion singledrop mikroextraction) Disperzní mikroextrakce z kapaliny do kapaliny (Dispersive liquid liquid microextraction) Extrakce pomocí jednorázových špiček pipet (Disposable pipette tips extraction) Eluční činidlo Elektoronová ionizace (Electron ionization) Evropská zdravotnická organizace (European Medicines Agency) Ionizace elektrosprejem (Electrospray ionization) Kyselina mravenčí (Formic acid) Lipoproteiny s vysokou hustotou (High density lipoproteins) Vícenásobná extrakce z kapaliny do kapaliny pomocí dutého vlákna (Hollow fiber liquid liquid liquid microextraction) Mikroextrakce do kapaliny pomocí dutého vlákna (Hollow fibre liquid phase microextraction) Hydrofilní interakční kapalinová chromatografie 9
10 HMG-CoA HPLC i.d. ICH IS IT LC-MS/MS LDL LLE LLOQ LOD LOQ m/z MALDI ME MeOH MEPS MIPs MS MS/MS NA o-oh AT PČ PDMS PE PLE (Hydrophilic Interaction Liquid Chromatography) 3-hydroxy-3-methylglutaryl-koenzymA Vysokoúčinná kapalinová chromatografie (High performance liquid chromatography) Vnitřní průměr (Inner diameter) Mezinárodní konference pro harmonizaci technických požadavků pro registraci humánních léčivých přípravků (The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use) Vnitřní standard (Internal standard) Iontová past (Ion Trap) Kapalinová chromatografie s tandemovou hmotnostní detekcí Lipoproteiny s nízkou hustotou (Low density lipoproteins) Extrakce kapaliny kapalinou (Liquid liquid extraction) Dolní mez kvantifikace (Lower limit of quantification) Limit detekce (Limit of detection) Limit kvantifikace (Limit of quantification) Poměr hmotnosti ku náboji Ionizace laserem za účasti matrice (Matrix-assisted laser desorption/ionization) Matricové efekty (Matrix effects) Methanol Mikroextrakce pomocí tuhého sorbentu (Microextraction by packed sorbent) Molekulárně vtištěné polymery (Molecularly imprinted polymers) Hmotnostní spektrometrie (Mass spectrometry) Tandemová hmotnostní spektrometrie (Tandem mass spectrometry) Not available (Není k dispozici) Orto-hydroxyatorvastatin Promývací činidlo Polydimethylsiloxan Účinnost procesu (Process efficiency) Tlaková kapalinová extrakce (Pressurized liquid extraction) 10
11 p-oh AT PP PTFE Q RAM RE RSD RV RV-d 6 RVL SBSE SDME SPE SPME SRM SST STD TDM TFC TOF UHPLC ULOQ UV VIS VLDL Para-hydroxyatorvastatin Precipitace proteinů (Protein precipitation) Polytetrafluorethylen Kvadrupól (Quadrupole) Materiály s omezeným přístupem (Restricted access materials) Výtěžnost (Recovery) Relativní směrodatná odchylka Rosuvastatin Deuteriem značený rosuvastatin Rosuvastatin lakton Sorpční extrakce pomocí magnetického míchadla (Stir bar sorptive extraction) Mikroextrakce pomocí jedné kapky (Single drop microextraction) Extrakce na tuhou fázi (Solid phase extraction) Mikroextrakce na tuhou fázi (Solid phase microextraction) Režim monitorování vybraných reakcí (Selected reaction monitoring) Test způsobilosti analytického systému (System suitability test) Standard Monitorování lékových hladin (Therapeutic drug monitoring) Kapalinová chromatografie s turbulentním průtokem (Turbulent-flow chromatography) Analyzátor doby letu (Time of Flight) Ultra vysokoúčinná kapalinová chromatografie (Ultra high performance liquid chromatography) Horní limit kvantifikace (Upper limit of quantification) Ultrafialová oblast Viditelná oblast Lipoproteiny o velmi nízké hustotě (Very low density lipoproteins) 11
12 1. Úvod Statiny jsou řazeny mezi hypolipidemika, neboli látky snižující hladinu cholesterolu v krvi. Mechanismus jejich účinku spočívá v inhibici klíčového enzymu endogenní syntézy cholesterolu, a to 3-hydroxy-3-methylglutarylkoenzym A reduktázy. Cholesterol je obsažen v krvi ve formě lipoproteinů a jeho hladina zvýšená nad 5 mmol/l vede k vyššímu riziku aterosklerózy a jejím komplikacím. Rosuvastatin a atorvastatin byly pro tuto práci vybrány vzhledem k jejich výhodným vlastnostem, z nichž je důležitý dlouhý biologický poločas, který umožňuje podání kdykoliv během dne. Tím se liší od ostatních statinů, které musí být podány večer kvůli jejich farmakokinetice, protože syntéza cholesterolu je nejvyšší během noci. Výhodou rosuvastatinu je i nízké ovlivnění izoenzymů CYP-450, a tím i méně nežádoucích účinků. Atorvastatin a rosuvastatin byly doposud stanovovány především kapalinovou chromatografií ve spojení s ultrafialovou detekcí či s tandemovou hmotnostní spektrometrií. V současné době jsou upřednostňovány rychlejší metody analýzy, proto je hojně využívána UHPLC-MS/MS. Tato metoda umožňuje jednak rychlou, ale i citlivou analýzu. Biologické vzorky těchto statinů byly obvykle upravovány extrakcí kapaliny kapalinou (LLE), či extrakcí na tuhou fázi (SPE). Tyto metody jsou metodami invazivními z důvodu potřeby relativně velkého množství biologického materiálu (vzorku). MEPS metoda na rozdíl od těchto umožňuje využít velmi malé množství vzorků i rozpouštědel, což snižuje invazivnost metody a činí metodu ekonomičtější ve srovnání s předchozími. 12
13 2. Cíl a zadání práce Tato diplomová práce se zabývá stanovením atorvastatinu a rosuvastatinu pomocí ultra vysokoúčinné kapalinové chromatografie ve spojení s tandemovou hmotnostní spektrometrií typu trojitého kvadrupólu a úpravou biologického vzorku (séra) s obsahem těchto dvou hypolipidemik. Nejprve budou změřena hmotnostní spektra rosuvastatinu, rosuvastatinu laktonu, N-desmethylrosuvastatinu, atorvastatinu, atorvastatinu laktonu, p-hydroxy a o-hydroxy atorvastatinu a vybrány prekurzory. Poté budou nastaveny podmínky pro SRM přechody pro jednotlivé analyty, a to pozorovací čas, napětí na vstupním kuželu. Následně bude provedena MS/MS analýza a zvoleny jednotlivé fragmenty, pro které bude nastavena kolizní energie. Po nastavení těchto podmínek bude ověřena linearita, citlivost a opakovatelnost metody s použitím vnitřních izotopicky značených standardů. Předchozí podmínky budou využity pro optimalizaci úpravy biologického vzorku séra pomocí metody MEPS (mikroextrakce pomocí tuhého sorbentu). Bude optimalizováno složení promývacího a elučního činidla, použitého pro extrakci analytů ze séra a bude zvolena vhodná tuhá fáze pro sorpci analytů. Po optimalizaci extrakční metody bude tento postup zvalidován dle směrnic ICH (Mezinárodní konference pro harmonizaci technických požadavků pro registraci humánních léčivých přípravků). 13
14 3. Teoretická část 3.1 Statiny Statiny jsou nejvýznamnější skupinou léčiv, která se používá ke snižování hladiny cholesterolu v krvi. Jejich účinek vychází z blokády enzymu zvaného 3- hydroxy-3-methylglutaryl-koenzym A reduktáza, čímž dojde k zastavení syntézy cholesterolu v organismu. Tato hypolipidemika jsou proto primárně využívána pacienty pro snížení hladiny cholesterolu. Důsledkem snížení hladiny cholesterolu v krvi je snížení kardiovaskulární mortality. První statin, který byl objeven a izolován z houby Penicillium citrinum v roce 1976 japonským profesorem Endem, mevastatin, nebyl v praxi využit. Následně byly získávány další statiny syntetickou nebo semisyntetickou cestou a v klinické praxi již našly uplatnění. Statiny lze rozdělit do dvou skupin. První skupinou statinů jsou proléčiva, která jsou podávána ve formě laktonové. Do této skupiny je řazen simvastatin a lovastatin, které se v organismu mění na vlastní účinnou formu, t.j. formu kyseliny. Druhou skupinu tvoří látky ve formě vlastní účinné kyseliny, kam patří rosuvastatin, atorvastatin, fluvastatin a pravastatin [1][2][3] Struktura statinů Většina statinů obsahuje ve své molekule částečně hydrogenované naftalenové jádro, kterým se váže na hydrofobní část receptoru. Na toto základní stavební jádro je navázán 2-methylbutanoylový zbytek a dále pomocí dvou uhlíkatého spojovacího můstku seskupení, které je podobné strukturou kyselině mevalonové. Tato část molekuly je tedy zodpovědná za inhibici syntézy cholesterolu. Ke statinům s touto strukturou je řazen mevastatin, lovastatin, pravastatin či simvastatin [4]. Základní aromatickou kostrou rosuvastatinu (Obr. 1) je pyrimidin. Rosuvastatin jako jediný z užívaných statinů obsahuje polarizovanou methylsulfonamidovou skupinu, díky níž má nejvyšší počet vazebných interakcí s HMG-CoA reduktázou a největší schopnost tento enzym blokovat. Navíc je díky této struktuře vysoce hydrofilní a je selektivně vychytáván jaterními buňkami, což je pozitivní vzhledem k nežádoucím účinkům především na příčně pruhované svalstvo [5]. Mezi metabolity rosuvastatinu se řadí rosuvastatin lakton (Obr. 2) a N-desmethyl rosuvastatin (Obr. 3). 14
15 Obr. 1: Struktura rosuvastatinu Obr. 2: Struktura rosuvastatinu laktonu Obr. 3: Struktura N-desmethylrosuvastatinu V případě atorvastatinu (Obr. 4) tvoří aromatickou část skeletu pyrrol, který je složitě substituovaný aromatickými cykly. Metabolity atorvastatinu jsou atorvastatin lakton (Obr. 5) a orto-/para-hydroxyatorvastatin (Obr. 6 a Obr. 7) [4]. Obr. 4: Struktura atorvastatinu Obr. 5: Struktura atorvastatinu laktonu 15
16 Obr. 6: p-hydroxyatorvastatin Obr. 7: o-hydroxyatorvastatin Fyzikálně chemické vlastnosti analyzovaných statinů Rosuvastatin, atorvastatin a jejich deriváty jsou bílé krystalické látky, které nejsou rozpustné ve vodě. Rozpouští se v polárních organických rozpouštědlech, jako je acetonitril. Název Sumární vzorec Molekulová hmotnost log P pka Rosuvastatin C 22 H 28 FN 3 O 6 S 481,54 0,892 4,25 Rosuvastatin lakton C 22 H 26 FN 3 O 5 S 463,52 1,202 13,22 N-desmethyl rosuvastatin C 21 H 26 FN 3 O 6 S 467,51 0,740 4,25 Atorvastatin C 33 H 35 FN 2 O 5 558,64 3,846 4,29 Atorvastatin lakton C 33 H 33 FN 2 O 4 540,62 3,902 13,39 o-hydroxy atorvastatin C 33 H 35 FN 2 O 6 574,64 4,103 4,29 p-hydroxy atorvastatin C 33 H 35 FN 2 O 6 574,64 3,210 4,29 Tab. 1: Fyzikálně chemické vlastnosti atorvastatinu, rosuvastatinu a jejich metabolitů [6][7] Mechanismus účinku statinů Mechanismus účinku statinů spočívá v blokádě klíčového enzymu dráhy syntézy cholesterolu. Tato hypolipidemika se naváží na enzym HMG-CoA reduktázu, čímž zabrání přeměně HMG-CoA na mevalonát a dojde k zastavení endogenní syntézy cholesterolu především v hepatocytech. Snížením nitrobuněčné tvorby cholesterolu jsou buňky nuceny zvýšit tvorbu LDL-receptorů na svém povrchu, což vede k poklesu LDL (low density lipoproteins) cholesterolu v krvi. V závislosti na dávce zároveň dochází k poklesu triacylglyceridů v krvi a ke vzestupu HDL (high density lipoproteins) cholesterolu [8][9]. 16
17 3.1.4 Farmakologické účinky a klinické využití statinů Účinky inhibitorů HMG-Co A reduktázy můžeme rozdělit na lipidové a nelipidové. Mezi lipidové účinky řadíme snížení LDL cholesterolu, VLDL cholesterolu, a tím pádem také snížení celkové hladiny cholesterolu v krvi vlivem zablokování syntézy cholesterolu, dále snížení triacylglyceridů a zvýšení hladiny HDL cholesterolu. Blokací tvorby dílčího produktu syntézy cholesterolu, mevalonátu, je také ovlivněn vznik dalších působků v těle, které se podílí na metabolismu a přenosu buněčného signálu. Z tohoto důvodu statiny neovlivňují pouze syntézu cholesterolu, ale ovlivňují také endotelovou dysfunkci, zánětlivé reakce, migraci a proliferaci buněk hladkého svalstva stěny cév. Proto jsou využívány k léčbě kardiovaskulárních onemocnění, jejich primární i sekundární prevenci. Podle nových studií se ukazuje, že by tyto jejich nelipidové účinky mohly mít i pozitivní vliv na rozvoj osteoporózy, Alzheimerovy choroby či diabetu mellitu druhého typu [9][10]. Mezi nežádoucí účinky statinů patří myalgie a myopatie. Tyto nežádoucí účinky jsou spojené se svalovou únavou, bolestí, slabostí a křečemi. Ve vzácných případech může dojít k rhabdomyolýze, kdy se do krevního řečiště uvolní myoglobin, což může způsobit poškození dalších orgánů, především ledvin, kde je myoglobin v tubulech vychytáván. Symptomy rhabdomyolýzy jsou nevolnost, zvracení, svalová bolest, teplota a tmavě zbarvená moč. Toto onemocnění lze diagnostikovat pomocí vyšetření svalového enzymu kreatininkinázy a myoglobinu v séru. O něco častější je zvýšení hodnot jaterních testů, bolesti hlavy, svědění, dyspeptické potíže a exantém [9][11][12] Metody stanovení rosuvastatinu a atorvastatinu Rosuvastatin a atorvastatin jsou nejčastěji stanovovány pomocí kapalinové chromatografie s UV/VIS či MS detekcí. Obvykle jsou stanoveny z lidské plazmy či moči nebo z krysí a psí plazmy. Pro analýzu atorvastatinu je využívána i metoda plynové chromatografie (GC), u které je třeba pro citlivá stanovení provést dvojnásobnou derivatizaci vzorku. Později byla vyvinuta metoda využívající kapalinovou chromatografii s UV či hmotnostní detekcí. V současné době se využívají k detekci i detektory diodového pole (DAD) nebo například fluorescenční detekce. Vzorky obsahující atorvastatin jsou nejčastěji upravovány pomocí extrakce kapaliny kapalinou (LLE Liquid liquid extraction) nebo 17
18 extrakce na tuhou fázi (SPE solid-phased extraction). Analytické kolony jsou nejčastěji typu C18 a mobilní fáze tvoří polární směsi organických rozpouštědel, a to zejména acetonitrilu (ACN), methanolu (MeOH), kyseliny mravenčí (FAc Formic Acid). Délka analýzy se liší podle použité metody a bývá přibližně v rozmezí dvou až několika desítek minut. Pro správnou detekci a stanovení analytů jsou využívány vnitřní standardy analyzovaných látek (IS). V současné době jsou pro hmotnostní detekci nejvhodnějšími standardy izotopicky značené analyty. Citlivost analýz se pohybuje v rozmezí od 0,5 ng/ml až řádově k desetinám mikrogramů na mililitr [1][13]-[18]. Rosuvastatin je nejčastěji stanovován z lidské plazmy (Tab. 2). Vzorky jsou upravovány metodami LLE a SPE. Délka kolony pro LC analýzu se pohybuje v širokém rozmezí, typem sorbentu bývá nejčastěji silikagel s kovalentně navázanými ligandy C18. Mobilní fáze je složena z polárních rozpouštědel, kde směsi nejčastěji obsahují vodu, MeOH, ACN, FAc nebo octan amonný (AmAc Amonium Acetate). Během analýzy je třeba zachovávat ph v rozmezí 4,0-5,0, protože při těchto hodnotách nedochází ke konverzi laktonové a kyselé formy statinů, ke které dochází při ph vyšším i nižším, než je uvedené rozmezí. Díky vhodně zvolenému ph dochází k zachování poměru množství laktonové formy a kyseliny ve vzorku. Proto je ph používaných pufrů a aditiv velmi důležité a během celé analýzy a ve všech krocích úpravy vzorků udržované v daném rozmezí. Při stanovování rosuvastatinu kapalinovou chromatografií je nejčastěji k detekci využita tandemová hmotnostní spektrometrie. Analýza probíhá obvykle v intervalu 2-20 minut. Citlivost analýz je v rozmezí 0,02 1,0 ng/ml [19]- [32]. 18
19 Stanovovaná látka Původ vzorku Způsob úpravy Analytická kolona Mobilní fáze Detekce Prekurzorový iont Čas analýzy (min) Citlivost Reference rosuvastatin N-desmethylrosuvastatin plazma SPE XBridge C18 (50 mm x 2,1 mm, 5 μm) gradientová eluce A: H 2 0 B: 0,005% hydroxid amonný v MeOH LC-MS/MS ESI [M-H] - NA 0,100 ng/ml 0,0500 ng/ml [19] rosuvastatin IS = ketoprofen krysí plazma LLE Kromasil KR100-5C A (250 mm x 4,6 mm, 5 μm) isokratická eluce 0,05M FAc:ACN (55:45) UV (240 nm) 15 min 0,2 ng/ml [20] atorvastatin o-hydroxyatorvastatin p-hydroxyatorvastatin IS = rosuvastatin lidská plazma LLE Symmetry C18 (100 mm x 4,6 mm, 5 μm) isokratická eluce 0,03% FAc:ACN (30:70) LC-MS/MS ESI + 2 min 100 pg/ml [21] [M+H] + rosuvastatin fenofibrát IS = karbamazepin lidská plazma LLE X-Terra MS C-18 (50 mm x 4,6 mm, 5,0 μm) isokratická eluce 0,05M FAc:ACN (45:55) LC-MS/MS ESI + 5 min 0,1 ng/ml [22] [M+H] + rosuvastatin IS = rosuvastatin-d 6 lidská plazma SPE Luna C18 (150 mm x 4,6 mm, 5 μm) Luna C18 (50 mm x 2,0; 1,0; 0,5 mm, 3 μm) gradientová eluce A: H 2 0 B: MeOH: H 2 0 (95:5) + 0,2% FAc LC-MS/MS 4 min NA [23] rosuvastatin IS = atorvastatin lidská plazma SPE YMC J' Sphere ODS H-80 (150 mm x 4,6 mm, 4,0 μm) isokratická eluce 0,2% FAc v H 2 0 a ACN (40:60) LC-MS/MS ESI + 6 min 1,0 ng/ml [24] [M+H] + rosuvastatin IS = hydrochlorothiazid lidská plazma LLE Zorbax XDB-C18 (150 mm x 4,6 mm, 5 μm) isokratická eluce MeOH:voda (75:25, ph 6 pomocí vodného roztoku amoniaku) LC-MS/MS ESI 3 min 0,020 ng/ml [25] rosuvastatin gemfibrozil IS = celekoxib lidská plazma LLE X-Terra C18 (150 mm x 4,6 mm, 5,0 μm) isokratická eluce 0,01M AmAc:ACN: MeOH (50:40:10) UV (275 nm) 20 min 0,03 μg/ml 0,30 μg/ml [26] 19
20 Stanovovaná látka rosuvastatin IS = rosuvastatin-d 6 rosuvastatin lakton IS = rosuvastatin lakton d 6 atorvastatin IS = atorvastatin-d 5 Atorvastatin lakton-d pitavastatin rosuvastatin IS = rosuvastatin-d 3 N-desmethyl rosuvastatin IS = N- desmethylrosuvastatin-d 6 pitavastatin rosuvastatin pitavastatin lakton rosuvastatin IS = estron rosuvastatin IS = cilostazol Původ vzorku Způsob úpravy SPE lidská plazma SPE moč lidská plazma SPE lidská plazma LLE lidská plazma LLE Analytická kolona Luna PhenylHexyl (150 mm x 2 mm, 3,0 μm) ACQUITY UPLC BEH HILIC (50 mm x 2,1 mm, 1,7 μm) Shim-pack VP-ODS (150 mm x 4,6 mm, 5 μm) Phenomenex Luna C18 (2) 5 μm (150 mm x 4,6 mm) Mobilní fáze isokratická eluce A: 0,1% FAc v H 2 0 B: MeOH (25:75) gradientová eluce A: 2,5 mm bikarbonát amonný v H 2 0:ACN:hydroxid amonný (98:2:0,01) B: 2,5 mm bikarbonát amonný v H 2 0:ACN:hydroxid amonný (2:98:0,01) MeOH: H 2 0:FAc (75:25:0,05) Atlantis C18 column (150 mm x 2,1 mm, 5 μm) H 2 0 s 0,2% FAc: MeOH (30:70) Detekce Prekurzorový iont LC-MS/MS ESI + [M+H] + LC-MS/MS ESI - [M-H ]- LC-MS/MS ESI + [M+H] + Čas analýzy (min) 15 min 2 min 6 min Citlivost NA [27] 0,100 ng/ml 0,08 ng/ml MeOH: 2% FAc v H 2 0 (80:20) LC-MS/MS 5 min 0,1 ng/ml [30] Reference [28] [29] LC-MS/MS + 5 min 0,2 ng/ml [31] [M+H] rosuvastatin rosuvastatin lakton IS = rosuvastatin-d 6 lidská plazma SPE Luna C18 (2) (150 mm x 4,6 mm, 5 μm) MeOH/ 0,2% FAc (70:30) LC-MS/MS + 3,5 min 0,1 ng/ml [32] [M+H] Tab. 2: Analytické metody pro stanovení rosuvastatinu. Vysvětlivky: ACN - acetonitril, AmAc - octan amonný, MeOH methanol, FAc kyselina maravenčí, IS vnitřní standard, LLE extrakce kapaliny kapalinou, SPE extrakce na tuhou fázi, NA not available, RV- rosuvastatin. 20
21 3.2 Ultra vysokoúčinná kapalinová chromatografie (UHPLC) UHPLC neboli ultra vysokoúčinná kapalinová chromatografie je relativně novým směrem, který dává nové možnosti ve využití kapalinové chromatografie. Tato separační metoda je založena na principu HPLC, která využívá kolon, kde velikost částic je menší než 2 mikrometry, optimálně okolo 1,7 µm, což umožňuje poskytnutí lepších výsledků separace, vyšší rozlišení, snížení meze detekce, zkrácení času analýzy, snížení objemů používaných vzorků a rozpouštědel. UHPLC instrumentace musí být konstruována tak, aby systém odolal zpětným vysokým tlakům, které jsou způsobeny použitou velikostí částic [33][34][35][36]. V současné době jsou využívány dva typy sorbentů. Prvním typem jsou silikagelové sorbenty, druhým hybridní fáze. Oba typy sorbentů mají své výhody. Silikagelové fáze jsou poměrné odolné, vykazují vysokou účinnost, ale nejsou stabilní v celém rozsahu ph. Hybridní sorbenty jsou mechanicky více odolné a díky spojení silikagelu s organickými polymery je možné tyto fáze využít v celém rozsahu ph. Oba typy fází mohou být modifikovány různými uhlovodíkovými ligandy- C18, C8, fenylem, kyanoskupinou a dalšími polárními skupinami. Moderní UHPLC začíná kromě reverzních a normálních fází využívat HILIC (hydrofilní interakční kapalinová chromatografie), iontovýměnné fáze, vícemodální fáze (mixed-mode) a další [33] [34][35][36]. Technika má široké využití především ve farmacii, toxikologii, metabolomice, genomice a v dalších odvětvích analýzy [33][34][35][36]. 3.3 Hmotnostní spektrometrie Hmotnostní spektrometrie (MS) je fyzikálně-chemická technika, která slouží k rozdělení látek po jejich ionizaci na základě poměru jejich hmotnosti a náboje m/z. MS je mimořádně citlivá, destruktivní metoda, která pro analýzu potřebuje minimum vzorku a kromě zjištění molekulové hmotnosti látky umožní získat i další informace o struktuře molekuly. Je to metoda, kterou lze využít pro kvalitativní i kvantitativní analýzu látek. Má široké uplatnění ve všech odvětvích analýzy [37][38]. Hmotnostní spektrometrie využívá hmotnostních spektrometrů, kde probíhají všechny základní kroky této metody: odpaření vzorku, ionizace, akcelerace iontů do hmotnostního analyzátoru, separace a detekce [37]. 21
22 3.3.1 Instrumentace MS Hmotnostní spektrometr se skládá ze tří základních částí: iontového zdroje, hmotnostního analyzátoru a detektoru [37]. Iontový zdroj Iontový zdroj slouží k převedení neutrálních částic na ionty. Ionizace může probíhat několika způsoby. Využívané ionizační techniky lze rozdělit na techniky měkké a tvrdé, podle toho, v jaké míře dochází k fragmentaci molekul. Mezi tvrdé techniky je řazena elektronová ionizace. Tato technika vyžaduje těkavé vzorky. Dochází k uvolnění valenčního elektronu vlivem interakce s volným elektronem či s valenčním elektronem ionizačního plynu a ke vzniku radikálové částice a dalších fragmentů [38][40]. Měkké ionizační techniky jsou využívány častěji a jsou šetrnější ke vzorku analyzované látky, protože nedochází v takové míře k fragmentaci molekuly. Vznikají kladné protonované či záporné deprotonované molekuly podle zvoleného pozitivního či negativního záznamu iontů. Mezi základní měkké ionizační techniky patří ionizace elektrosprejem (ESI), chemická ionizace za atmosférického tlaku (APCI), fotoionizace za atmosférického tlaku (APPI) a ionizace laserem za účasti matrice (MALDI) [38][40]. Ionizace elektrosprejem je založena na postupném odpařování rozpouštědla, které vede ke coulombické explozi a k rozpadu kapek na menší až k tvorbě desolvatovaných protonovaných iontů. Náboj na částice je vnesen ve formě napětí na kapiláru. APPI metoda je založená na interakci fotonu s prostředím analytu. K ionizaci tak dochází pomocí UV záření, vzniká protonovaná/deprotonovaná částice. APCI je založena na obdobném principu, pouze molekuly rozpouštědel jsou ionizovány koronovým výbojem. MALDI ionizace vychází z interakce matrice a laseru, kdy dochází k ionizaci daného prostředí, které dále interaguje s analytem a přenáší na něj vzniklý náboj [38][40]. Hmotnostní analyzátor Funkce hmotnostního analyzátoru spočívá v separaci iontů na základě poměru m/z. Separace probíhá za vysokého vakua. Mezi základní typy analyzátorů patří magnetický analyzátor, analyzátor doby letu (TOF), kvadrupólový analyzátor (Q), iontová past (IT) a Orbitrap. V magnetickém analyzátoru dochází k zakřivení dráhy letu 22
23 v závislosti na hodnotě m/z. TOF analyzátor rozdělí urychlené ionty podle různých rychlostí mimo magnetické pole v závislosti na jejich hmotnosti a náboji. ( Menší ionty letí rychleji a větší pomaleji ). Kvadrupólový analyzátor je složený ze 4 tyčí, na které je vkládáno stejnosměrné napětí. To společně s amplitudou a frekvencí předurčuje trajektorie drah iontů, po kterých se budou pohybovat podle hodnoty m/z. Při daném nastavení mají stabilní trajektorii pouze některé ionty o daném m/z a právě ty projdou analyzátorem k detektoru. Nastavení se postupně mění tak, aby prošly všechny ionty. Iontová past je trojrozměrnou analogií kvadrupólu. Orbitrap je typem detektoru, který je složen z vnější a středové elektrody, na něž je vloženo napětí. Analyzované ionty se pohybují podél a okolo středové elektrody a jsou děleny na základě různé frekvence harmonických oscilací [37][38][39]. Detektor Poslední základní částí hmotnostního spektrometru je detektor. Ten převádí proud iontů na proud elektronů, který vyrazí z detektoru elektron, jenž je následně zesílen zesilovačem (elektronásobiče), nebo foton, jehož signál je také zesílen (fotonásobiče), nebo dochází k vybití kondenzátoru sběrem elektronů na elektrodě (Faradayova klec). Po zesílení signálu je možné tento signál vyhodnotit [37][38]. Tandemová hmotnostní spektrometrie Tandemová hmotnostní analýza nebo také hmotnostní analýza n-tého stupně je založena na přítomnosti několika analyzátorů řazených sériově za sebou pomocí kolizní cely a slouží k bližší specifikaci látek. První analyzátor umožní rozlišení fragmentovaných iontů a volbu jednoho nebo více z nich, který je následně v kolizní cele podroben fragmentaci na produktové ionty, jenž jsou rozlišeny ve druhém analyzátoru [41]. 3.4 Spojení kapalinové chromatografie a hmotnostní spektrometrie Spojení kapalinové chromatografie a hmotnostní spektrometrie je v poslední době velmi rozšířenou analytickou technikou, která se rutinně používá v analytických laboratořích. Velkou výhodou spojení těchto dvou separačních technik je vysoká citlivost a selektivita, schopnost měřit více analytů současně a také krátká doba analýzy. 23
24 V minulosti bylo charakteristické náročnějším technickým spojením, protože zde byl velký rozdíl mezi tlakem v hmotnostním analyzátoru a v přiváděném vzorku. Navíc byly analyzované látky neseny v toku kapaliny, jejíž přebytek bylo třeba odstranit před vstupem do analyzátoru. Proto byly vyvinuty metody ESI, APCI, APPI, u kterých analýza probíhá za atmosférického tlaku a molekuly mobilní fáze se přímo účastní ionizačního procesu. V současné době je to plně rutinní spojení s řadou již zmíněných výhod [42][43]. 3.5 Způsoby úpravy vzorků Separační techniky jsou velmi často používány k analýze komplexních vzorků, jako je krev, sérum, plazma, moč či sliny. Analýza těchto složitých matric je využívána v mnoha oborech. Je základem pro monitorování lékových hladin (TDM) u pacientů, u kterých je třeba sledovat koncentraci léčiva v krvi, farmakokinetické studie, klinické studie či forenzní analýzu. Základním cílem úpravy vzorku je převést komplexní matrici na vzorek, který bude vhodný pro analýzu pomocí dané separační techniky. S rozvojem analytické chemie se příprava a úprava vzorků stává hlavní částí analytické metody, která zabere až 80 % celkového času kompletního rozboru vzorku [44]-[47]. Příprava vzorků se skládá z několika kroků izolace, přečištění a zakoncentrování analytů z matrice. Biologické vzorky nejsou pro LC-MS analýzu vhodné vzhledem k jejich komplexnímu složení. Obsahují velké množství interferujících sloučenin, mezi které patří soli, fosfolipidy a proteiny. Proteiny se mohou ireverzibilně navázat na tuhý sorbent, a tím snížit účinnost separační kolony a zvýšit zpětný tlak, popřípadě mohou ucpat fritu, kolonu či dávkovač. Z toho důvodu jsou v bioanalýze často používány předkolony. Navíc fosfolipidy a soli mohou způsobit matricové efekty, tedy potlačení či zvýšení signálu analyzované látky [45]. Techniky pro přípravu vzorků lze rozdělit do dvou základních skupin (Obr. 8). Do skupiny základních konvenčních technik je možné zařadit precipitaci proteinů (PP), extrakci kapaliny kapalinou (LLE) a extrakci na tuhou fázi (SPE). Druhou skupinu tvoří moderní metody. Tyto moderní metody by měly umožnit využití malého objemu vzorků a rozpouštědel a úsporu času. Do druhé skupiny lze zařadit on-line metody, vysoce selektivní a mikroextrakční techniky [45]. 24
25 Obr. 8: Rozdělení technik pro úpravu biologických vzorků: PP precipitace proteinů. LLE Extrakce kapaliny kapalinou. SPE - Extrakce na tuhou fázi. RAM - Materiály s omezeným přístupem. TFC Kapalinová chromatografie s turbulentním průtokem. SDME Mikroextrakce pomocí jedné kapky. HF- LPME - Mikroextrakce kapalnou fází pomocí dutého vlákna. DLLME - Disperzní mikroextrakce z kapaliny do kapaliny. SBSE Sorpční extrakce pomocí magnetického míchadla. DPX - Extrakce pomocí jednorázových špiček pipet. MEPS - Mikroextrakce pomocí plněného tuhého sorbentu. SPME - Mikroextrakce na tuhou fázi. MIP s - Molekulárně vtištěné polymery Konvenční techniky Konvenční techniky pro úpravu vzorků jsou stále velmi široce používány ve většině analytických laboratoří. Všechny metody jsou dobře reprodukovatelné, optimalizovatelné a snadno se mohou automatizovat. Z toho důvodu není vývoj a ověření metody problematické. Jsou jednoznačné, spolehlivé a zaručují dosažení správných a přesných výsledků v přiměřeném čase [45]. Srážení proteinů (Precipitace proteinů PP) Srážení proteinů (Precipitace proteinů) odstranění proteinů ze vzorku je široce rozšířená metoda, která využívá srážecí reakce bílkovin s organickými rozpouštědly, anorganickými solemi, či silnými kyselinami. Problémem této metody může být změna ph při použití silných kyselin, pokud je stabilita analytů závislá na ph, což může 25
26 negativně ovlivnit výsledky následného stanovení. Použité organické rozpouštědlo musí být kompatibilní s použitou mobilní fází LC. Další komplikací mohou být zbylé složky použité matrice, které způsobují vznik matricových efektů a interferujících píků v chromatogramu. Výhody této metody spočívají v jednoduchosti provedení, časové nenáročnosti metody a snadné optimalizaci [44][45][48]. Provedení PP spočívá v několika krocích (Obr. 9). Po přidání srážecího činidla dochází k reakci mezi proteinem a precipitační přísadou. Vzniká precipitát, který je třeba odstranit, například pomocí centrifugace či filtrace. Získaný supernatant se dále může upravit, nebo přímo zanalyzovat. Obr. 9: Postup precipitace. 1. Zkumavka s matricí. 2. Přidání precipitačního činidla. 3. Probíhající precipitační reakce. 4. Vznik precipitátu a supernatantu. Extrakce kapaliny kapalinou (LLE) Extrakce kapaliny kapalinou je další metoda přípravy vzorku, kterou řadíme mezi konvenční techniky. Je to jedna z prvních metod, která byla používána pro úpravu biologických vzorků. LLE je založena na přechodu jednoho nebo více analytů mezi dvěma vzájemně nemísitelnými kapalinami. Jednu kapalnou fázi tvoří nepolární organické rozpouštědlo, druhým rozpouštědlem je rozpouštědlo polární, kterým může být vodný roztok nebo polární organické rozpouštědlo. Analyt je obvykle součástí roztoku s polárním rozpouštědlem. Přechod analytu mezi fázemi je dán jeho rozpustností v použitých rozpouštědlech. Jeho hydrofilní či hydrofobní vlastnosti jsou vyjádřeny pomocí rozdělovacího koeficientu oktanol-voda n, který je vyjádřen poměrem koncentrací analyzované látky v oktanolové c(o) a vodné c(v) fázi (Obr. 10). n= c(o) c(v) Obr. 10: Vzorec pro výpočet rozdělovacího koeficientu. 26
27 Jestliže je rozdělovací koeficient látky vyšší než jedna, pak má analyt lipofilní charakter, pokud je menší než jedna, vykazuje vlastnosti hydrofilní. Rozpouštědla používaná pro LLE jsou seřazena dle relativní permitivity a rostoucí polarity v eluotropní řadě rozpouštědel. Nejčastěji užívaná polární a nepolární rozpouštědla jsou uvedena v následující tabulce (Tab. 3). Kromě polarity rozpouštědla je extrakce ovlivněna fyzikálně chemickými vlastnostmi analytu, poměrem rozpouštědel a dobou trvání extrakce. Rozpouštědlo Relativní permitivita ε Rozpustnost ve vodě (g/l) Pentan 1,8 0,04 Hexan 1,9 0,14 Cyklohexan 2,0 0,10 Tetrachlormethan 2,2 0,80 Toluen 2,4 0,47 Benzen 2,3 1,80 Diethylether 4,3 74,2 Chloroform 4,8 10,0 Aceton 20,7 mísitelný Dioxan 2,2 mísitelný 1-Pentanol 13,9 21,9 Tetrahydrofuran 7,6 mísitelný Ethylacetát 6,0 86,0 Diethylamin 3,6 mísitelný Acetonitril 37,5 mísitelný 1-Butanol 17,1 79,0 Pyridin 12,4 mísitelný Isopropylalkohol 19,9 mísitelný Ethanol 24,3 mísitelný Methanol 32,6 mísitelný Ethylenglykol 37,7 mísitelný Kyselina octová 6,1 mísitelný Tab. 3: Eluotropní řada rozpouštědel [49]. Postup LLE (Obr. 11, Obr. 12) spočívá v přidání rozpouštědla opačné polarity k matrici, protřepání a odstředění vzorku a oddělení fáze, ve které se analyt nachází. Vzorek je následně možné upravit, nebo rovnou analyzovat pomocí separačních metod. Extrakce kapaliny kapalinou je metoda, která je stále široce využívaná. Je selektivní a snadno automatizovatelná, ale má několik nevýhod, a to časovou náročnost, 27
28 vysokou spotřebu organických rozpouštědel, a tím i značný vznik toxického odpadu. Není vhodná pro polární látky, což metabolity léčiv často bývají [33][45][49]-[53]. Obr. 11: Provedení LLE [54]. 1) 2) 3) Obr. 12: Princip extrakce:1) Přidání nemísitelné fáze k biologickému roztoku. 2) Protřepání. 3) Oddělení fází a analytu podle afinity k rozpouštědlům. Extrakce na tuhou fázi (SPE) Extrakce na tuhou fázi (SPE) je velmi rozšířená a v současné době velmi populární metoda úpravy biologických vzorků. Kromě přečištění vzorku a izolace analytu je tato technika využívána také pro zakoncentrování analyzované látky. Princip SPE spočívá v distribuci analyzované látky mezi kapalnou fázi a tuhý sorbent. Pro zachycení analytu na tuhém sorbentu musí mít analyty dostatečnou afinitu k danému povrchu. Adsorpce analytu na tuhou fázi je závislá na polárních, nepolárních a iontových interakcích mezi separovanou látkou a tuhým sorbentem [45][46][55]-[58]. Základem pro extrakci pomocí tuhé fáze je volba správného a vhodného sorbentu, která závisí na fyzikálně chemických vlastnostech analytu, typu matrice a na interakcích sorbentu a analytu. Používané typy tuhé fáze jsou obdobné jako u kapalinové chromatografie. Mezi obvyklé sorbenty užívané při SPE patří tuhé fáze s chemicky modifikovaným silikagelem (C8, C18), grafitovým uhlíkem, iontovýměnné materiály, polymery, vícemodální sorbenty (fáze, které obsahují nepolární funkční skupinu a kationtový, nebo aniontový měnič) a monolitické sorbenty. Pro selektivní extrakci na tuhou fázi jsou používány sorbenty, které obsahují imunoafinitně vázanou protilátku, nebo MIP s (molecularly imprinted polymers). Imunosorbenty jsou využívány především v medicíně, biologii a v potravinářství. Obsahují protilátku navázanou na podkladu, která je specifická pro zjišťovaný analyt. Sorbent je obvykle umístěn v plastových nebo skleněných kolonkách anebo je slisován se skleněnými vlákny či polytetrafluorethylenem do disků [45][46][55]-[58]. 28

29 Extrakce na tuhý sorbent se provádí v několika krocích. Nejprve je třeba aktivovat kolonku s tuhou fází vhodným rozpouštědlem, aby došlo k otevření vazebných míst pro analyt. Tento krok se nazývá kondicionace kolonky a provádí se rozpouštědlem, které je složením blízké vzorku. Následně je biologický vzorek nanesen na kolonu, kde dojde k zachycení analytu. Po adsorpci analyzované látky je možné sorbent promýt vhodným promývacím roztokem, aby došlo k odstranění balastních látek. Poté se analyt vymyje z tuhé fáze vhodným elučním činidlem a převede se tak do roztoku, který je možné analyzovat kapalinovou chromatografií (Obr. 13). Obr. 13: Postup SPE. 1. Kondicionace kolonky. 2. Nasátí vzorku. 3. Promytí. 4. Vysušení. 5. Eluce analytu [59]. SPE se oproti předchozím metodám vyznačuje mnoha výhodami. Je to efektivní metoda s nízkou spotřebou toxických organických rozpouštědel. Poskytuje přesné a spolehlivé výsledky, je snadno automatizovatelná a jednoduchá na provedení. Nevýhodou může být časová náročnost SPE a cena vybavení pro extrakci [45][46][55]-[58] Moderní techniky Moderní metody, které jsou v současné době často využívány, vychází z předchozích konvenčních metod. Zavádění nových metod pro úpravu vzorků má velký význam především z důvodu zrychlení a zkvalitnění analýzy. Tyto techniky redukují počet kroků úpravy vzorku, umožňují selektivnější izolaci analytu, využívají nižší objemy vzorků (nejsou tak invazivní pro pacienty) a rozpouštědel a také snižují cenu celkové úpravy vzorku. Jsou ovšem náročnější na instrumentální vybavení 29
30 laboratoře a vyžadují určitou zkušenost analytika. V následující části práce jsou uvedeny některé moderní metody úpravy vzorků biologické matrice [45][46]. Mikroextrakce vycházející z LLE Mikroextrakční metody vycházející z extrakce kapaliny kapalinou jsou založené na principu LLE, a to na rozdílné afinitě analytu k použitým rozpouštědlům. Mezi tyto mikroextrakční metody patří SDME (extrakce pomocí jedné kapky), HF-LPME (mikroextrakce kapalnou fází pomocí dutého vlákna), DLLME (disperzní mikroextrakce z kapaliny do kapaliny). Tyto metody jsou nejčastěji využívány ve spojení s rychlou a vysoce účinnou kapalinovou chromatografií. Jejich výhodou je především velké snížení objemů používaných rozpouštědel, možnost velkého zakoncentrování analytu a použití malého množství matrice [45]. SDME mikroextrakce pomocí jedné kapky (Single-drop microextraction) Extrakce pomocí jedné kapky byla vyvinuta jako technika pro zakoncentrování analytu. Využívá malého objemu organického rozpouštědla nemísitelného s vodnou fází (1-10 μl) k extrakci analyzované látky. Těkavé látky jsou extrahovány do rozpouštědla umístěného v kapce visící na jehle a kapka je následně zanalyzována [33][60][61][62]. HF-LPME mikroextrakce do kapaliny pomocí dutého vlákna (Hollow Fiber Liquid-phase micorextraction) Při této extrakci se využívá duté vlákno, což je porézní membrána, která nese organické rozpouštědlo nemísitelné s vodou umístěné ve stěnách pórů. To musí být s vláknem kompatibilní. Po ponoření vlákna do vodného roztoku analytu dochází k extrakci dané látky stejným způsobem jako u LLE. V některých případech (dynamická HF-LLME) je vlákno umístěno na jehle, pomocí které se extrakce provádí. Variantou této mikroextrakce je třífázová extrakce HF-LLLME (vícenásobná etrakce z kapaliny do kapaliny pomocí dutého vlákna), kdy po extrakci do organického rozpouštědla nemísitelného s vodou je provedena zpětná extrakce do vodné fáze uvnitř vlákna [33][62]. 30
31 DLLME disperzní mikroextrakce kapaliny kapalinou (Dispersive liquid-liquid microextraction) Metoda disperzní mikroextrakce je založena na třísložkovém systému rozpouštědel, který tvoří vodná fáze, nepolární rozpouštědlo nemísitelné s vodou (extrakční činidlo) a polární s vodou mísitelné rozpouštědlo (dispergující činidlo). Kapky extrakčního roztoku jsou dispergované do vodné fáze a odpovídající směs obou fází je potom vstřikována do vody. Po následné centrifugaci či ochlazení částice extrakčního činidla sedimentují v kónické zkumavce. Výsledná usazená fáze je mikrostříkačkou odebrána a aplikována do analyzátoru (nejčastěji plynové chromatografie). Klíčovou roli v této metodě má disperzní roztok, který společně s extrakčním činidlem tvoří kapičky ve vodné fázi a tvoří až 99 % celkového objemu vzorku. Výhodou této metody je její jednoduchost, nízká cena, vysoká výtěžnost a krátká doba extrakce [33][62][63]. Mikroextrakce vycházející z SPE Metody vycházející z extrakce na tuhou fázi jsou založeny na obecném principu, tzn. zachycení analytu z kapalné fáze na tuhou vlivem interakce analytu s tuhým sorbentem. Některé z těchto mikroextrakcí mají velký potenciál stát se konvenčními technikami pro úpravu vzorků pro rychlou analýzu vzorků pomocí UHPLC. Toto platí především pro techniku MEPS. Dále do této skupiny řadíme SPME (mikroextrakci na tuhou fázi), DPX (mikroextrakce pomocí jednorázových pipetovacích špiček), SBSE (sorpční extrakce pomocí magnetického míchadla). MEPS mikroextrakce pomocí plněného tuhého sorbentu (Microextraction by packed sorbent) MEPS je nová vysoce citlivá a selektivní metoda využívající principu SPE. SPE a SPME se od metody MEPS liší uložením sorbentu a jeho množstvím. V SPE je sorbent (množství okolo 50 mg) uložen v kolonce, v SPME ve formě vlákna v jehle a v metodě MEPS je sorbent (1-4 mg) uložen ve formě patrony, která je součástí jehly (Obr. 14). Provedení metody MEPS je stejné jako u metody SPE (Obr. 13). Výhodou metody je použití velmi malého množství vzorku i rozpouštědel (obvykle 10 µl). MEPS může být ale použita i pro analýzu větších množství (až 1000 µl). Větší objemy se analyzují odebíráním alikvotních podílů 100 µl nebo 250 µl. Sorbenty pro MEPS mohou být typu reverzních fází, normálních fází, smíšené či 31
32 iontovýměnné. Mezi často používané tuhé fáze se řadí sorbenty s modifikovaným silikagelem (C8, C18), RAM, MIPs, polystyren-divinylbenzen kopolymerem. Extrakce se provádí buď manuálně, pomocí ruční automatické stříkačky (evol, SGE)(Obr. 14), nebo autosamplerem. Obr. 14: A) evol(sge) automatická [64]. B) MEPS manuální stříkačka [65]. Metoda je používána pro úpravu biologických vzorků, především moči, plazmy, séra a krve. MEPS má velký potenciál pro využití v UHPLC/MS analýze, především díky její rychlosti, jednoduchému provedení, nízké spotřebě toxických rozpouštědel i biologických vzorků. Nevýhodou této metody může být pořizovací cena sorbentů a ruční stříkačky. Tento působ úpravy vzorků je využíván jak v bionalýze, tak ve farmaceutickém průmyslu a v dalších odvětvích ve spojení s kapalinovou a plynovou chromatografií, kapilární elektroforézou a s dalšími separačními metodami [33][65]- [68]. SPME mikroextrakce na tuhou fázi (Solid phase microectraction) Metoda mikroextrakce na tuhou fázi (Obr. 15) je miniaturizovanou metodou SPE, kterou je možné dále rozdělit na dvě skupiny: na SPME pomocí vlákna (Fiber SPME) a kapilární SPME (viz 3.3.2). SPME pomocí vlákna je metoda založena na využití speciální stříkačky, jejíž součástí je jehla z nerezové oceli. Uvnitř jehly je uloženo vlákno potažené organickým sorbentem. Toto vlákno je vysunováno a zasunováno do stříkačky pomocí pístu. Sorbentem může být PDMS (polydimethylsiloxan), polyakryláty, polyethylenglykol, divinylbenzen a další 32
33 sloučeniny. Tuhá fáze umožňuje adsorpci a desorpci analytu v závislosti na použitých rozpouštědlech. Metoda se využívá v bioanalýze vzorků krve, plazmy, moči. Objem vzorku nutný pro analýzu je menší než 1 ml, což je jednou z výhod metody. Dalšími pozitivy je jednoduchost metody a velmi nízká spotřeba rozpouštědel [33][69][70]. Obr. 15: Postup SPME: 1. Stříkačka s vláknem a vialka. 2. Proniknutí stříkačky do vialky. 3. Vysunutí vlákna. 4. Adsorpce analytů na tuhý sorbent. 5. Zasunutí vlákna do stříkačky. 6. Vlákno připravené pro separační analýzu v jehle [71]. DPX - extrakce pomocí jednorázových špiček pipet (disposable pipette tips) Metoda DPX (Obr. 16) je extrakční technika, která využívá tuhého sorbentu uloženého v jednorázových špičkách pipet mezi dvěma fritami. Sorbent je tak ihned po nasátí vzorku smísen s roztoky analytů, aniž by bylo třeba použít dalšího rozpouštědla. Sníží se tím množství použitého rozpouštědla a je umožněna rychlejší extrakce. Analyt lze dávkovat přímo pipetou [46][72]. Obr. 16: Špička se sorbentem pro DPX [72]. 33
34 SBSE sorpční extrakce pomocí magnetického míchadla (stir bar sorptive extraction) SBSE je extrakční metoda, která se od SPME liší v uložení extrakční fáze. V tomto případě je umístěna na magnetickém míchadle. Mezi nejčastěji používané tuhé fáze patří PDMS. Tato technika úpravy vzorků se používá k izolaci organických látek o nízké koncentraci z vodné fáze, kde je přechod analytu založen na rozdělovacím koeficientu oktanol-voda (viz LLE). Postup spočívá ve vložení míchadla do vialky, kde se otáčí a adsorbuje na sebe analyt. Následně je vyjmuto a po zahřátí je analyzovaná látka uvolněna a lze ji dále detekovat pomocí plynové chromatografie (GC) i chromatografie kapalinové, u které není třeba termální desorpce. Zahřátí míchadla je umožněno vlastnostmi PDMS, který tvoří sorpční fázi. Tento povrch je možné použít i při vysokých teplotách. Metoda SBSE má využití v biologické, farmaceutické a v potravinové analýze látek [33][73][74]. Vysoce selektivní metody pro úpravu vzorku Vysoce selektivní metody jsou metody, které využívají specifických reakcí analyzovaných látek s určitými činidly či molekulami. Mezi tyto techniky řadíme techniku MIPs molekulárně vtištěné polymery (molecularly imprinted polymers), imunoafinitní SPE a aptamery. MIPs molekulárně vtištěné polymery (Molecularly imprinted polymers) MIPs jsou selektivní materiály, které jsou používány pro extrakci na tuhou fázi. Molekulárně vtištěné sorbenty jsou připravovány třemi metodami metodou kovalentního vtištění, nekovalentního vtištění či kombinací dvou předchozích. Vtištěné komplexy jsou tvořené z monomerů, které byly podrobeny polymerizaci. Výsledné otisky mají stérickou a chemickou paměť a mohou tak navázat strukturně stejné či velmi podobné jednotky. Výhodami této techniky jsou selektivita, stabilita a jednoduchost provedení. Nevýhodou je dlouhý čas extrakce, použití velkých objemů rozpouštědel a riziko degradace polymerů vlivem teplotních změn [33]. Imunoafinitní SPE (Immunoaffinity SPE) Imunoafinitní extrakce je založena na interakcích mezi antigenem a protilátkou. Imunosorbent je složen z protilátek specifických pro daný analyt ukotvených na tuhé 34
35 fázi. Látky o molekulární hmotnosti menší než 1000 Da musí být navázány na hapten, protože jinak by nevyvolaly žádnou odpověď. Protilátky musí být inertní vůči sorbentu, na kterém jsou navázány, dále musí být inertní chemicky i biologicky, jednoduše aktivovatelné a nesmí vykazovat nespecifické reakce. Výhodou této metody je opět její jednoduché provedení a vysoká selektivita [69][75]. Aptamery Aptamery jsou oligonukleotidy, které mohou s vysokou specifitou a selektivitou vázat velké množství různých molekul, například drogy, proteiny a jiné organické i anorganické molekuly. Metody využívající aptamery jsou stále předmětem výzkumu a vývoje [33][76]. Online techniky pro úpravu vzorku Kapilární SPME (In-tube solid phase extraction) Kapilární SPME je automatizovaná verze mikroextrakce na tuhou fázi, která může být jednoduše použita ve spojení s HPLC systémem pro on-line úpravu vzorku. Je velmi vhodná pro úpravu biologických vzorků, a to především moči, séra a plazmy. Tato metoda je označena termínem kapilární (in-tube), protože sorbent, na kterém probíhá vlastní extrakce, tvoří povlak vnitřní stěny křemenné kapiláry na rozdíl od konvenční SPME, kde je na jejím povrchu. Křemenná kapilára je v systému umístěna na stejném místě jako dávkovací smyčka v běžném autosampleru. V průběhu analýzy je vzorek opakovaně vstřikován skrz tuto kapiláru, kde dojde k sorpci analytu. Ten je následně vymyt a unášen vhodnou mobilní fází na analytickou kolonu HPLC či UHPLC systému a dále analyzován. Výhodou metody je redukce času analýzy, vyšší přesnost, rychlost, nulová spotřeba rozpouštědel, nízká spotřeba vzorku a vysoká citlivost díky zakoncentrování vzorku [33][77][78]. RAM materiály s omezeným přístupem (Restricted access materials) Metoda využívající materiály s omezeným přístupem je dalším typem technik, které lze využít k on-line úpravě vzorků v přímém spojení s HPLC systémem. Umožňuje přímé vstřikování biologické tekutiny do systému. Tyto sorbenty prezentují speciální třídu materiálů, které se využívají k rozdělení biologických vzorků na proteinovou matrici a analyty. Proces je založený na rozdílné molekulové hmotnosti a velikosti částic. Nízkomolekulární složky jsou schopné proniknout do pórů a zachytit 35
36 se v sorbentu. Vysokomolekulární složky, jako jsou proteiny, nukleové kyseliny a další, se na materiál nezachytí díky speciálnímu povrchu tvořenému hydrofilními skupinami, které znemožňují jejich navázání, a také díky omezené velikosti pórů v sorbentu. Tato technika úpravy vzorků je využívána k analýze moči, séra či plazmy v bioanalýze. Jako sorbenty se obvykle používají hydrofobní materiály (C4, C8, C18) [79][80]. TFC - Kapalinová chromatografie s turbulentním průtokem (Turbulent-flow chromatography) Kapalinová chromatografie s turbulentním průtokem je další metodou, která se využívá ve spojení s kapalinovou chromatografií a s hmotnostním detektorem při analýze biologických vzorků. Princip této metody spočívá v separaci malých molekul analytu od vysokomolekulární matrice pomocí nízkých difuzních koeficientů proteinů. Separační účinnost je důsledkem turbulencí v mobilní fázi. Turbulentní průtok je možné přeformovat použitím vhodných kolon. Kolony v TFC jsou krátké, úzké a jejich náplň je tvořena velkými částicemi, jejichž typický rozměr je μm. Zadržené analyty jsou potom z kolony vymyty vhodnou organickou mobilní fází a unášeny na analytickou kolonu, kde jsou dále separovány[33][73][80][81]. 3.6 Validace metody a matricové efekty Validace analytické metody Validace metody je série experimentů, kterými lze zjistit nejdůležitější charakteristiky metody a potvrdit reprodukovatelnost, spolehlivost výsledků a vhodnost pro zamýšlené použití. Cílem validace je vyšetřit hranice, ve kterých je zkušební postup použitelný, a zajistit, aby při opakovaném použití v jedné nebo více laboratořích metoda poskytovala správné výsledky [82]. Aby výsledky byly spolehlivé při každém použití metody, musí být podmínky přesně definovány, nebo musí být stanovena kritéria, která umožní spolehlivé použití analytického systému. Tato kritéria se obecně nazývají test způsobilosti analytického systému SST (System suitability test). U chromatografických metod musí být zajištěna účinnost a separační schopnost. Dvěma základními údaji jsou požadavky na opakovatelnost a rozlišení dvou vybraných píků. Případně lze určit počet teoretických pater, faktor symetrie (rozmezí 0,8-1,5 pro léčivé látky pokud není v lékopise uvedeno jinak), retenční čas a retenční poměr stanovované látky. Mezi faktory, které mohou 36
37 ovlivnit chromatografické chování, je zahrnuto složení mobilní fáze, její ph, iontová síla, průtoková rychlost, délka kolony, teplota, tlak, typ stacionární fáze včetně velikosti částic a porozity. Pro léčiva je maximální relativní směrodatná odchylka ploch hlavního píku pro opakované nástřiky uvedena v tabulce a nesmí překročit danou hodnotu (Tab. 4) [82][83]. Počet jednotlivých nástřiků B (%) Maximální relativní směrodatná odchylka 2,0 0,41 0,59 0,73 0,85 2,5 0,52 0,74 0,92 1,06 3,0 0,62 0,89 1,10 1,27 Tab. 4: Maximální relativní směrodatné odchylky pro opakované nástřiky předepsaného porovnávacího roztoku dle ČL B horní limit daný v definici jednotlivých lékopisných článků minus 100 % za předpokladu, že horní limit je určen podle reprodukovatelnosti metody [83]. Obecně analytické parametry ověřované při validaci metod jsou selektivita, linearita, rozsah, přesnost (opakovatelnost, reprodukovatelnost), správnost, limit detekce a limit kvantifikace, robustnost, výtěžnost a stabilita analytu v matrici (Obr. 17) [82][84]. Selektivita metody udává schopnost změřit správně a specificky analyt v přítomnosti složek, které mohou být součástí vzorku (např. metabolity, nečistoty, matricové součásti). Linearita je schopnost metody dávat výsledky přímo úměrné koncentraci analytu ve vzorku. Rozsah je interval mezi dvěma koncentracemi látky, kdy je metoda lineární a dává přesné a správné metody. Limit detekce (LOD) je nejnižší detekovatelná koncentrace analytu, která není hodnocena kvantitativně. Limit kvantifikace (LOQ) je nejmenší množství analytu, které lze stanovit s danou přesností a správností. Je možné takto určit maximální (ULOQ) a minimální (LLOQ) koncentraci látky, kterou lze danou metodou ještě stanovit. Přesnost metody je míra shody jednotlivých výsledků metody, která je opakovaně prováděna s homogenním vzorkem. Je vyjádřena směrodatnou odchylkou. U přesnosti metody rozlišujeme opakovatelnost (metoda je prováděna jedním analytikem na jednom přístroji, se stejnými činidly a jedním vzorkem) a reprodukovatelnost (různá laboratoř, jiný analytik, různá činidla a přístroje, stejný vzorek). Správnost je odchylka zjištěné hodnoty od referenční hodnoty, která se zjistí jinou ověřenou metodou, nebo analýzou modelového vzorku se známým obsahem standardu. Vyjadřuje se jako výtěžnost (recovery v procentech), 37
38 kterou lze vypočítat jako podíl stonásobku zjištěné hodnoty a hodnoty skutečné [82]. Robustnost metody je míra schopnosti metody podat správné a přesné výsledky, i když dojde k menším změnám pracovních podmínek při daném pracovním postupu. Stabilita je důležitá vlastnost analytu. Aby látka zůstala stabilní, musí být dodržovány specifické podmínky uchovávání v závislosti na vlastnostech této látky [82][84]. Obr. 17: Shrnutí validačních parametrů podle EMA (Evropská zdravotnická organizace) a ICH [45]. * koncentrační hladina x počet opakování., X parametr není vyžadován, - parametr je vyžadován. Kvantitativní hodnocení metody je obvykle prováděno pomocí vhodného vnitřního standardu. Tato metoda spočívá v přídavku známého množství látky-vnitřního standardu k neznámé koncentraci zjišťovaného analytu, nebo v porovnání množství neznámé látky s kalibrační křivkou standardů o známé koncentraci. Jako vnitřní standardy jsou užívány různé typy látek. Nejvhodnější pro LC-MS analýzy se jeví použití izotopicky značených standardů, které mají stejné vlastnosti jako. Pro každý analyt by měla být jako vnitřní standard zvolena totožná molekula, která je izotopicky značená [37] Matricové efekty Matricové efekty je možné definovat jako změny účinnosti ionizace, které jsou způsobené přítomností látek, jež vstupují do iontového zdroje společně s látkou analyzovanou. Matricové efekty mohou signál analyzované látky potlačovat nebo zesilovat. Přesný mechanismus vzniku matricových efektů není znám, ale 38
39 pravděpodobně vychází z interakce mezi analytem a současně eluovanou látkou. Efekty mohou být způsobeny endogenními složkami vzorku, které nejsou odstraněny během úpravy vzorku (soli, polární látky fenoly, aminy, uhlovodíky, peptidy, lipidy, a další), použitými chemikáliemi (soli, pufry, organické kyseliny, iont-párové látky), či může dojít k vnesení reziduí polymerů či dalších balastních látek během extrakčního postupu. Matricové efekty jsou kromě toho ovlivněny typem dané matrice, typem analytu, způsobem úpravy vzorku, separací a detekcí [85][86][87]. Matricové efekty lze určit třemi způsoby. Metodou post-kolonové infúze, metodou postextrakčního přídavku a metodou porovnání směrnic kalibračních křivek. Při post-kolonové infúzi je na kolonu nastřikován extrakt vzorku bez analyzovaných látek a analyt je vstřikován v konstantním množství do mobilní fáze pomocí t-kusu za kolonou. Endogenní sloučeniny z matrice potom mohou způsobit změnu intenzity signálu. Celý proces probíhá za stejných podmínek jako vlastní analýza. Metoda post-extrakčního přídavku vychází z porovnání naměřených hodnot dvou různých vzorků čisté mobilní fáze, matrice upravené optimalizovanou metodou a následně obohacené o analyt (Obr. 18). Obr. 18: Metoda post-extrakčního přídavku. A) Čistá mobilní fáze. B) Matrice upravená optimalizovanou metodou následně obohacená o analyt. C) Matrice s obsahem analytu upravená optimalizovanou metodou. Výpočet matricových efektů (ME), výtěžnosti metody (RE) a účinnosti procesu (PE) pomocí ploch pod křivkou jednotlivých typů vzorků. [85]. Třetí způsob určení matricových efektů spočívá v porovnání směrnic standardní kalibrační křivky a matricové kalibrační. Při potlačení signálu vlivem matricových efektů dojde ke zmenšení směrnice kalibrační křivky matricové, při zesílení signálu k nárůstu směrnice matricové kalibrační křivky [85][86][88]. 39
40 Matricové efekty lze eliminovat či snížit několika různými způsoby v jednotlivých krocích úpravy a analýzy vzorku (Tab. 5)[85]. redukce ME Analytické metody Příprava vzorku důkladné přečištění zředění vzorku vyšší selektivita SPE, RAM, LLE MIP, imunoafinitní SPE, RAM Chromatografie vyšší separační účinnost změna selektivity HILIC, MF, SF gradientová eluce Hmotnostní spektrometrie vyšší selektivita ionizační techniky méně náchylné záznam negativních iontů APPI, APCI, EI-MS k ME Kalibrace a kvantifikace vhodný kalibrační postup IS, metoda přídavku standardu, matricová kalibrační křivka Validace určení ME, správnosti a přesnosti metody Tab. 5: Eliminace či snížení matricových efektů [85]. SPE extrakce na tuhou fázi, RAM materiály s omezeným přístupem, LLE Extrakce kapaliny kapalinou, MIP Molekulárně vtištěné polymery, APPI fotoionizace za atmosférického tlaku, APCI chemická ionizace za atmosférického tlaku, EI-MS hmotnostní detekce za použití elektronové ionizace, IS vnitřní standard. 40
41 4. Experimentální část 4.1 Materiál a pomůcky Chemikálie Atorvastatin (čistota 98%), Toronto Research Chemicals, Kanada Atorvastatin-d5 (čistota 99%), Toronto Research Chemicals, Kanada Atorvastatin lakton (čistota 98%), Toronto Research Chemicals, Kanada p-hydroxyatorvastatin (čistota 98%), Toronto Research Chemicals, Kanada o-hydroxyatorvastatin (čistota 95%), Toronto Research Chemicals, Kanada Rosuvastatin (čistota 97%), Toronto Research Chemicals, Kanada Rosuvastatin-d6 (čistota 98%), Toronto Research Chemicals, Kanada Rosuvastatin lakton (čistota 95%), Toronto Research Chemicals, Kanada N-desmethylrosuvastatin (čistota 98%), Toronto Research Chemicals, Kanada ultračistá voda vyrobena na Farmaceutické fakultě v Hradci Králové, Mili-Q, Millipore, před použitím byla přefiltrována acetonitril acetonitrile, LC-MS Chromasolv, minimum 99,9%, Sigma-Aldrich kyselina octová acetic acid, minimumn 99,9%, Sigma-Aldrich amoniak amonnium hydroxide solution pro LC-MS, minimum 25% ve vodě, Sigma-Aldrich. Sérum LYO HUM N 10x5, Erba Lachema s.r.o. Brno Přístroje a pomůcky MEPS stříkačka o kapacitě 500 μl (SGE Analytical Science) Sorbent C8, C18, M1 MEPS BIN pro XCHANGE Ruční automatická evol pipeta (SGE Analytical Science) Automatické pipety se špičkami BIOHIT (FISCHER SCIENTIFIC, ČR) Plastové stříkačky a PTFE filtry 4 mm x 0,2 μm, ISO-DISC Filtres (Supelco Analytical) Acquity UPLC systém (Waters) o Čerpadlo o Automatický dávkovač o Kolonový termostat Analytická kolona Acquity BEH C18 (50 2,1 mm, 1,7 μm, Waters) 41
42 Hmotnostní analyzátor typu trojitého kvadrupólu Quattro Micro (Waters) Software MassLynx Minitřepačka: Ika MS 3 basic (IKA WORKS, USA) Ultrazvuková vodní lázeň Sonorex Digital (Bandelin, Německo) Laboratorní ph metr (HANNA Instruments, Itálie) Magnetická míchačka: (IKA big squid, USA) Analytické váhy: Sartorius 2004 MP (SARTORIUS, Německo) 4.2 Příprava roztoků Příprava zásobních roztoků Pro přípravu zásobních roztoků rosuvastatinu, N-desmethylrosuvastatinu, deuteriem značeného rosuvastatinu, atorvastatinu laktonu, p-hydroxyatorvastatinu, o- hydroxyatorvastatinu a deuteriem značeného atorvastatinu bylo jako rozpouštědla využito mobilní fáze, jejíž složení odpovídalo počátku gradientu, tj. acetonitril: 0,5 mm octan amonný ph 4,0 v poměru 30:70. Pro přípravu zásobních roztoků rosuvastatinu laktonu a atorvastinu byl použit stoprocentní acetonitril. K lepšímu rozpuštění látek byla použita ultrazvuková lázeň a třepačka. Zásobní roztoky byly skladovány v lednici. Jejich koncentrace byla 10-3 mol/l Příprava pracovních roztoků Pracovní roztoky byly připravovány ředěním zásobních roztoků mobilní fází odpovídající složením počátku gradientu (viz výše) v koncentracích mol/l vždy před každým měřením. Pracovní roztok směsi standardů byl připraven odebráním 100 µl ze zásobního roztoku každého standardu a doplněním objemu do 1 mililitru mobilní fází, jejíž složení odpovídalo počátku gradientu (viz výše). Takto byl získán roztok směsi standardů o koncentraci 10-4 mol/l. Dále docházelo k ředění této směsi na koncentrace v rozmezí mol/l pomocí mobilní fáze o složení odpovídajícímu počátku gradientu (viz výše). Stejným způsobem byla připravena směs deuteriem značených vnitřních standardů, a to buď v celém rozmezí kalibrační křivky, nebo směs o koncentraci mol/l. 42
43 4.2.3 Příprava mobilní fáze Mobilní fáze pro ředění roztoků byla připravena smísením roztoku acetonitrilu a 0,5 mm octanu amonného o ph 4,0 v poměru odpovídajícímu počátku gradientu, tj. 30:70. 5 mm octan amonný o ph 4,0 byl připraven odměřením 71,3 µl kyseliny octové do 250 ml kádinky s 200 ml vody. Tento roztok byl následně titrován ředěným hydroxidem amonným na požadované ph 4,0. Roztok byl kvantitativně převeden do 250,0 ml odměrné baňky a vodou doplněn po rysku. Roztoky byly připravovány každý den čerstvé. 0,5 mm octan amonný o ph 4,0 byl připraven naředěním roztoku 5 mm octanu amonného o ph 4,0. 10 ml 5 mm octanu amonného o ph 4,0 bylo převedeno do odměrné baňky a doplněno vodou po rysku. 4.3 Optimalizace UHPLC-MS podmínek Pro chromatografickou analýzu s hmotnostní detekcí byla použita již dříve vyvinutá a zvalidovaná metoda, o které pojednává diplomová práce Pavla Svobody [89]. Byla použita analytická kolona Acquity BEH C18 (50 x 2,1 mm, 1,7 µm, Waters), gradientová eluce s použitím mobilní fáze ACN:0,5 mm octan amonný ph 4,0 o průtoku 0,3 ml/min a nástřik 5 μl. Pro hmotnostní detekci byla použita ionizační technika ionizace elektrosprejem v negativním i pozitivním módu ESI - /ESI +. Iontový zdroj byl nastaven v ESI takto: napětí na kapiláře 2,5 kv, teplota iontového zdroje 130 C, extrakční kužel 1 V, hexapól 0,5 V. Zmlžujícím plynem byl dusík o průtoku 650 l/hod a teplotě 450 C. Dusík byl také použit na vstupním kuželu, a to o průtoku 70 l/hod. Napětí na vstupním kuželu (cone voltage) bylo nastaveno pro každý analyt zvlášť. Kvantifikace analytů byla provedena s využitím SRM (selected reaction monitoring) experimentu, kdy pro každý analyt byly vybrány ionty prekurzoru, fragmentu a byla optimalizována kolizní energie (Tab. 6). 43
44 ANALYTY ZPŮSOB IONIZACE PREKURZOROVÉ IONTY FRAGMENTY DWELL TIME CONE VOLTAGE KOLIZNÍ ENERGIE t R RV ESI- 479,8 418,2 0, ,28 DES RV ESI- 465,8 404,1 0, ,99 RV-d 6 ESI- 485,8 424,2 0, ,29 RVL ESI+ 463,9 270,0 0, ,57 AT-d 5 ESI- 562,1 283,3 0, ,24 p-oh-at ESI- 573,1 413,1 0, ,27 o-oh-at ESI- 573,1 278,3 0, ,90 AT ESI- 557,1 278,3 0, ,26 ATL ESI- 539,2 278,2 0, ,28 Tab. 6: Nastavení SRM přechodů pro jednotlivé analyty. 4.4 Optimalizace metody MEPS Pro úpravu biologického vzorku byla použita Metoda MEPS. Vzorek byl upravován pomocí MEPS stříkačky o kapacitě 500 µl (SGE Analytical Science). Jako sorbent byl použit sorbent C8 MEPS BIN pro XCHANGE. Bylo třeba zvolit vhodný sorbent, na který se analyty zachytí, dále promývací činidlo k odstranění nečistot z kolonky a eluční činidlo, které dostatečně vymývá analyty naadsorbované na kolonku Základní roztoky pro MEPS Základními roztoky pro přípravu promývacího a elučního činidla při metodě MEPS byly pufry 0,1 M a 0,01 M octanu amonného o ph 4,5. 0,1 M roztok octanu amonného byl připraven z 570,1 µl kyseliny octové, které se přidaly přibližně k 75 mililitrům vody v kádince. Následně byl roztok titrován vodným roztokem hydroxidu amonného na požadovanou hodnotu ph 4,5. Roztok byl následně kvantitativně převeden do 100 ml odměrné baňky. 0,01 M roztok octanu amonného o ph 4,5 byl připraven naředěním 10 ml 0,1 M roztoku octanu amonného o témže ph v odměrné baňce o objemu 100 ml. 44
45 4.4.2 Promývací činidlo Promývací činidlo sloužící k odstranění nečistot před vlastní elucí analytů bylo připraveno smísením acetonitrilu a 0,01 M roztoku octanu amonného o ph 4,5 v poměru 5:95. Při optimalizaci byly využity různé poměry těchto činidel Eluční činidlo Eluční činidlo využívané pro vymytí analytů z kolonky bylo připraveno smísením acetonitrilu a 0,1 M roztoku octanu amonného o ph 4,5 v poměru 95:5. Při optimalizaci byly využity různé poměry těchto činidel Příprava upravovaných vzorků Metoda byla nejprve vyvinuta a optimalizována na vzorcích standardů. Příprava jejich používané směsi je uvedena výše. Po optimalizaci podmínek byly metodou MEPS upravovány směsi analytů přidané do reálně matrice (sérum) s konstantním množstvím vnitřního standardu. Ty byly připravovány následujícím postupem (Tab. 7). Byly připraveny směsné roztoky standardů o koncentraci 10-5 mol/l a vnitřních standardů značených deuteriem o koncentraci mol/l (viz výše). V prvním kroku je přidáno 100 µl vnitřního standardu o dané koncentraci, v následujících již je 90 µl, protože zbylých 10 µl je přenášeno společně s analyty při ředění z koncentrovanějšího vzorku do směsi s koncentrací o řád nižší. Dochází tak k zachování koncentrace vnitřního standardu, která je mol/l. Koncentrace IS ve všech roztocích (1000 µl) 10-7 (1000 µl) 10-8 (1000 µl) 10-9 (1000 µl) (1000 µl) 100 µl 10-5 směsi STD µl IS µl plazmy 100 µl µl IS µl plazmy 100 µl µl IS µl plazmy 100 µl µl IS µl plazmy 100 µl µl IS µl plazmy (1000 µl) (1000 µl) (1000 µl) (1000 µl) 100 µl směsi STD µl IS µl plazmy 100 µl µl IS µl plazmy 100 µl µl IS µl plazmy 100 µl µl IS µl plazmy Tab. 7: Příprava matricové kalibrační křivky. 45
46 Takto připravené roztoky byly upraveny metodou MEPS dle postupu uvedeného v Tab. 8. Objem činidla Použité činidlo Aktivace kolonky 3x100 µl ACN Kondicionace kolonky 3x100 µl 0,1 M AmAc ph 4,5 Nanesení vzorku 50 µl Promytí 2x100 µl 0,1 M AmAc ph 4,5 Promytí 100 µl Promývací činidlo Eluce analytů 100 µl Eluční činidlo Promytí 100 µl Eluční činidlo Tab. 8: Postup úpravy vzorku metodou MEPS. Promývací činidlo - ACN:0,01 M AmAc 4,5 ph 5:95; Eluční činidlo ACN:0,1 M AmAc 4,5 ph 95:5. Eluce vzorku je prováděna nasátím 100 µl elučního činidla přes MEPS sorbent pomocí MEPS stříkačky a následně je eluát vnesen do plastové stříkačky s PTFE (polytetrafluorethylen) filtrem o velikosti pórů 0,20 μm, kde je přefiltrován a přenesen do 100 µl mikroinsertu. 4.5 Opakovatelnost metody v rámci SST Opakovatelnost metody byla ověřena po optimalizaci všech podmínek proměřením tří různých koncentračních hladin roztoku standardu sérií deseti po sobě jdoucích měření. Ta probíhají za stejných podmínek, na stejném přístroji a v krátkém časovém rozmezí. Opakovatelnost byla vyjádřena relativní směrodatnou odchylkou pro retenční čas a plochu píku všech analyzovaných látek. 4.6 Validace metody Na závěr byla metoda zvalidována v souladu s ICH směrnicí. Pro kvantitativní hodnocení byla využita metoda vnitřního standardu s využitím izotopicky značených IS. Byly ověřeny validační parametry, a to linearita, správnost, přesnost, matricové efekty, selektivita, rozsah, limit detekce a limit kvantifikace. Linearita byla ověřována pro všechny analyty pomocí standardní kalibrační křivky v rozsahu 0, nmol/l. V tom samém rozsahu byla ověřena také linearita matricové kalibrační křivky. Správnost a přesnost metody byla ověřena na třech koncentračních hladinách (5, 50 a 500 nmol/l pro všechny analyty) během třech po sobě následujících dní. K ověření matricových efektů a správnosti metody byly použity dva vzorky pro každý analyt, a to 46
47 čisté sérum a blankové sérum upravené metodou MEPS a obohacené analyty (viz odstavec 3.6.2). Selektivita byla ověřena proměřením blanku (čistého séra upraveného metodou MEPS) a porovnáním naměřených chromatogramů s chromatogramy analyzovaných látek. Tímto způsobem bylo ověřeno, že v séru nejsou obsaženy látky, které by byly eluovány ve stejných retenčních časech jako analyty. Roztoky, které byly použity ke zvalidování metody, byly připraveny postupy uvedenými výše (Tab. 7). 47
48 5. Výsledky a diskuze 5.1 Výchozí podmínky metody UHPLC-MS/MS Separace a detekce analytů probíhala na základě již vyvinuté a optimalizované metody, o které pojednává Diplomová práce Pavla Svobody [89] UHPLC separace Pro separaci analytů byl využit UHPLC systém z důvodu vysoké separační účinnosti a rychlé analýzy. Byla použita analytická kolona Acquity BEH C18 (50 x 2,1 mm, 1,7 µm, Waters). Mobilní fázi tvořila směs ACN a 0,1 mm AmAc o ph 4,0, byla použita gradientová eluce s mobilní fází o počátečním složení 30:70. Podmínky pro UHPLC separaci jsou shrnuty v tabulce (Tab. 9, Tab. 10). Aditivum octanu amonného o ph 4,0 bylo zvoleno z toho důvodu, že v rozmezí ph 4,0-5,0 nedochází ke konverzi laktonové a kyselé formy statinů. Analýza statinů touto metodou trvala 7,5 minuty, což je velkou výhodou metody. Ukázkový chromatogram všech analytů je uveden na Obr. 19. Podmínky pro UHPLC separaci Analytická kolona kolona Acquity BEH C18 (50 x 2,1 mm, 1,7 µm, Waters) Mobilní fáze ACN a 0,1 mm AmAc o ph 4,0 Počátek gradientu mobilní fáze 30:70 Průtok 0,3 ml/min Nástřik 5 μl Tab. 9: Podmínky UHPLC separace. Program gradientové eluce Čas [min] Octan amonný ACN Gradientová křivka 0, , , , Tab. 10: Gradientová eluce - program. 48
49 Obr. 19: Záznam SRM přechodů pro standardní roztok analytů o koncentraci mol/l [90]. 49
50 5.1.2 Tandemová hmotnostní detekce Pro detekci analytů byla zvolena tandemová hmotnostní spektrometrie. Jako analyzátor byl použit hmotnostní analyzátor typu trojitého kvadrupólu. Pro ionizaci látek byl využit iontový zdroj typu elektrospreje v negativním i pozitivním módu ESI - /ESI +. Nastavení iontového zdroje je shrnuto v tabulce (Tab. 11). Nastavení elektrospreje napětí na kapiláře 2,5 kv teplota iontového bloku 130 C extrakční kužel 1 V hexapól 0,5 V sušící plyn dusík o průtoku 650 l/hod a o teplotě 450 C vstupní kužel dusík o průtoku 70 l/hod napětí na vstupním kuželu každý analyt zvlášť (Tab. 6) Tab. 11: Základní nastavení MS detektoru. Pro každý analyt byl nejprve vybrán vhodný prekurzorový iont a fragment v ESI, který dával nejcitlivější odezvu, a ten byl využit pro nastavení SRM přechodů (Tab. 6). Příklady výběru fragmentového a prekurzorového iontu pro jednotlivé analyty jsou znázorněny na Obr
51 51
52 Obr. 20: MS a MS/MS spektrum iontů v ESI negativním módu pro a)at, b) RV [90], d) RV-de, e) RV-d 6, f) ATL, g) o-ohat, h) p-ohat, i) AT-d 5 ; v pozitivním módu pro c) RVL. 52
53 5.2 Použití vnitřních standardů Pro správné provedení kvantitavní analýzy je třeba k hodnocení využít vnitřních standardů. V této diplomové práci byly použity dva izotopicky značené standardy. Rosuvastatin značený deuteriem (RV-d 6 ) a atorvastatin značený deuteriem (AT-d 5 ). RV-d 6 byl použit jako referenční látka pro rosuvastatin, N-desmethylrosuvastatin. AT-d 5 byl použit pro rosuvastatin lakton, atorvastatin a všechny jeho zjišťované metabolity. IS pro jednotlivé analyty nebyly použity z důvodu jejich vysoké ceny. 5.3 Optimalizace MEPS Pro vývoj a optimalizaci metody MEPS byla použita jako výchozí bod již dříve vyvinutá metoda, která sloužila pro analýzu atorvastatinu a jeho metabolitů (Tab. 12). Z této metody byly převzaty základní kroky pro úpravu biologického vzorku i jednotlivá použitá rozpouštědla a metoda byla dále optimalizována. Pro zjišťované analyty bylo třeba vybrat vhodný sorbent, eluční a promývací činidlo tak, aby výtěžnost metody byla pro každý analyt co nejvyšší. Objem činidla Použité činidlo Kondicionace kolonky 3x100 µl ACN Kondicionace kolonky 3x100 µl 0,1 M AmAc ph 4,5 Nanesení vzorku 50 µl Promytí 100 µl 0,1 M AmAc ph 4,5 Promytí 100 µl Promývací činidlo (ACN: 0,01 M AmAc 15:85) Eluce analytů 100 µl Eluční činidlo (ACN-0,1M AmAc 95:5) Promytí 100 µl Eluční činidlo Tab. 12: Výchozí metoda pro stanovení AT a jeho metabolitů [91] Výběr sorbentu a optimalizace elučního činidla Pro zachycení analytů ze vzorku bylo třeba vybrat vhodný sorbent. Byly zkoušeny tři typy sorbentů, a to sorbent C8, C18 a M1. Dále bylo třeba optimalizovat eluční činidlo, které je používáno k vymytí analyzovaných látek ze sorbentu. Jako eluční činidlo byla používána směs acetonitrilu a roztoku 0,1 M octanu amonného ph 4,5. ph 4,5 bylo zvoleno z důvodu potlačení interkonverze laktonové formy statinů 53
54 a formy kyseliny. Směs byla zkoušena v různých poměrech těchto dvou složek z důvodu různé polarity analytů. Byly zaznamenány odezvy na hmotnostním spektrometru a vypočteny výtěžnosti. Během volby vhodného sorbentu byly vždy zkoušeny tři vzorky, které byly upraveny za použití dané tuhé fáze, a výsledky byly srovnávány se standardem, to znamená s množstvím analytu v neupraveném vzorku. První ze zkoušených směsí byla směs acetonitrilu a octanu amonného v poměru 85:15. Nejlepší výsledky vykazoval sorbent C18, kde se výtěžnost pohybovala okolo 100% pro všechny analyty. Na ostatních sorbentech potom v rozmezí % (Obr. 21) C8 C18 M1 RV RV-de RVL Obr. 21: Výtěžnost pro jednotlivé sorbenty za použití EČ v poměru ACN:pufr 85:15. Dalším zkoušeným elučním činidlem byla směs ACN a 0,1 M AmAC ph 4,5 v poměru 90:10. Výtěžnost metody pro EČ v poměru 90:10 se pohybovala okolo hodnoty 100% pro rosuvastatin a desmethylrosuvastatin, pro rosuvastatin lakton v rozmezí % (Obr. 22) C8 C18 M1 RV RV-de RVL Obr. 22: Výtěžnost pro jednotlivé sorbenty za použití EČ v poměru ACN:pufr 90:10. 54
55 Posledním elučním činidlem, které bylo testováno na všech třech typech sorbentů, byla směs ACN a 0,1M AmAc ph 4,5 v poměru 95:5. Výtěžnost metody pro poslední eluční činidlo byla ve velmi úzkém rozpětí pro všechny použité sorbenty. Nejlepší se jevila u sorbentu C8 a C18, kde se pohybovala v obdobném rozmezí %. U sorbentu M1 byla v porovnání se zbylými dvěma sorbenty nízká pro desmethylrosuvastatin (Obr. 23) C8 C18 M1 RV RV-de RVL Obr. 23: Výtěžnost pro jednotlivé sorbenty za použití EČ v poměru ACN:pufr 95:5. Po srovnání výtěžností všech tří sorbentů při použití jednotlivých směsí elučního činidla bylo vybráno nejlepší eluční činidlo (Tab. 13, Tab. 14). Jako nejvhodnější se po provedených analýzách jevilo použití směsi ACN a 0,1M AmAc o ph 4,5 v poměru 95:5, protože toto eluční činidlo mělo nejlepší hodnoty výtěžností v procentuálním rozmezí ve srovnání s ostatními dvěma činidly. Sorbent C8 Sorbent C18 Sorbent M1 85:15 90:10 95:5 85:15 90:10 95:5 85:15 90:10 95:5 RV 85,9 101,3 103,3 101,6 99,4 101,2 97,7 102,6 98,6 RV-de 81,3 93,3 94,5 98,0 95,6 96,4 85,1 109,9 85,3 RVL 110,7 108,2 115,7 114,2 113,9 113,2 119,8 113,7 117,6 Tab. 13: Výtěžnosti pro RV a jeho metabolity za použití 3 typů zkoušených sorbentů a 3 směsí elučních činidel 55
56 Sorbent C8 Sorbent C18 Sorbent M1 85:15 90:10 95:5 85:15 90:10 95:5 85:15 90:10 95:5 RV 3,20 0,59 1,19 1,14 4,14 5,04 3,17 4,43 1,39 RV-de 2,98 0,53 0,82 4,45 3,10 2,90 1,24 4,69 2,19 RVL 2,25 0,68 0,85 1,31 2,84 2,33 1,79 3,16 1,79 Tab. 14: Relativní směrodadatné odchylky (% RSD) pro výtěžnost RV a jeho metabolity za použití 3 typů zkoušených sorbentů a 3 směsí elučních činidel. Po konečném výběru elučního činidla byly zhodnoceny výsledky pro jednotlivé sorbenty. Nejvhodnější tuhý sorbent nebyl ještě v této fázi optimalizace vybrán. Byl pouze vyloučen sorbent M1 z důvodu nízké hodnoty výtěžnosti pro desmethylrosuvastatin a největšímu rozpětí hodnot pro zkoumané analyty. V další fázi vývoje metody byly nadále použity dva sorbenty C8 a C Optimalizace promývacího činidla a konečná volba sorbentu Během této fáze optimalizace úpravy vzorku metodou MEPS byla zkoušena různá promývací činidla, která by byla nejvhodnější pro promytí a přečištění analytů naadsorbovaných na tuhý sorbent tak, aby ještě nedošlo k eluci analytů v tomto kroku. Byly zkoušeny různé poměry směsi ACN a 0,01M AmAc o ph 4,5. K eluci bylo použito již eluční činidlo v poměru 95:5 ACN a 0,1M AmAc ph 4,5 a byly testovány dva sorbenty C8 a C18. Prvním promývacím činidlem, které bylo zkoušeno, byla směs ACN a 0,01 M AmAc o ph 4,5 v poměru 20:80. Výtěžnost metody za použití tohoto promývacího činidla byla v dostačujícím rozmezí jak pro rosuvastatin, tak pro rosuvastatin lakton, ale velmi nízká pro desmethylrosuvastatin (Obr. 24) C8 C18 RV RV-de RVL Obr. 24: Výtěžnost pro jednotlivé sorbenty za použití EČ v poměru ACN:pufr 20:80. 56
57 Druhým promývacím činidlem, které bylo při optimalizaci testováno, byla směs stejných činidel, a to v poměru 15:85. Výtěžnost za použití tohoto promývacího činidla byla pro rosuvastatin na obou sorbentech okolo 100%, pro rosuvastatin lakton na sorbentu typu C8 také, na sorbentu C18 již byla vysoká, dosahovala hodnot nad 110%. Pro desmethylrosuvastatin byla výtěžnost vyšší za použití sorbentu C18 (zhruba 75%), při úpravě pomocí C8 dosahovala hodnot okolo 60% (Obr. 25) RV RV-de RVL 20 0 C8 C18 Obr. 25: Výtěžnost pro jednotlivé sorbenty za použití EČ v poměru ACN:pufr 15: 85. Třetím promývacím činidlem byla směs acetonitrilu a 0,01 M AmAc o poměru 90:10. Výtěžnost pro tuto směs promývacího činidla byla všechny analyty na obou sorbentech velmi podobná. (Obr. 26) RV RV-de RVL 20 0 C8 C18 Obr. 26: Výtěžnost pro jednotlivé sorbenty za použití EČ v poměru ACN:pufr 10:90. 57
58 Posledním zkoušeným promývacím činidlem byla směs ACN a 0,01 M AmAc o poměru 5:95. Výtěžnost této metody byla pro rosuvastatin na obou sorbentech velmi podobná, pohybovala se okolo hodnoty 90%. Pro desmethylrosuvastatin byla vyšší za použití sorbentu C8 (92%) a pro lakton rosuvastatinu na obou sorbentech dosahovala hodnot vyšších než 100%, při čemž nižší hodnota byla stanovena na sorbentu C18 (Obr. 27) RV RV-de RVL 20 0 C8 C18 Obr. 27: Výtěžnost pro jednotlivé sorbenty za použití EČ v poměru ACN:pufr 5:95. Po celkovém vyhodnocení výsledků u jednotlivých elučních činidel a sorbentů byl nakonec vybrán sorbent C8 a eluční činidlo ACN a 0,1 M AmAc o ph 4,5 v poměru 95:5. Vzhledem k volbě sorbentu bylo následně vybráno nejvhodnější promývací činidlo, kterým byl zvolen roztok ACN a 0,01 M AmAc ph 4,5 v poměru 5:95 z důvodu nejlepší výtěžnosti pro desmethylrosuvastatin, další dva porovnávané metabolity se svojí výtěžností příliš nelišily, proto bylo důležité zaměřit se na N- desmethylrosuvastatin. Sorbent C8 Sorbent C18 20:80 15:85 90:10 95:5 20:80 15:85 90:10 95:5 RV 95,3 102,0 89,4 90,1 94,0 97,7 91,5 90,3 RV-de 61,3 61,3 81,5 92,2 56,8 74,7 83,5 84,4 RVL 106,5 104,0 107,8 116,5 111,5 113,6 107,1 109,2 Tab. 15: Souhrn výtěžností jednotlivých analytů za použití sorbentů C8 a C18 a 4 různých promývacích činidel Následně byla tato metoda ověřena za použití biologického materiálu místo rozpouštědla a na směsi všech analytů včetně atorvastatinu a jeho derivátů. 58
59 Sorbent C8 Eluční činidlo ACN: 0,1 M AmAc ph 4,5 95:5 Promývací činidlo ACN: 0,01 M AmAc ph 4,5 5:95 Tab. 16: Konečná volba sorbentu a používaných činidel. 5.4 Ověření na biologickém materiálu Metoda optimalizovaná na standardních roztocích rosuvastatinu a jeho metabolitů rozpuštěných v mobilní fázi byla následně ověřena na vzorku s obsahem rosuvastatinu, ale i atorvastatinu včetně metabolitů obou základních látek, a to na směsi rozpuštěné v lidském séru. k sérovému prášku (lyofilizované standardní sérum). Sérum bylo získáno přidáním 5 ml destilované vody Byly proměřeny všechny zjišťované analyty, jejich vnitřní standardy a byly zaznamenány hodnoty ploch jednotlivých píků ve třech různých provedeních. Vzorky obsahovaly shodné množství analyzovaných látek a byly postupně upraveny dle optimalizovaného postupu MEPS (4.4 Optimalizace metody MEPS, Tab. 16). V této fázi byl navíc zaveden krok, kdy po nasátí elučního činidla musel být vzorek převeden do plastové stříkačky a přefiltrován z důvodu odstranění zbylých nečistot, které zůstaly i po použití metody MEPS. Z hodnot pro plochy píků jednotlivých analytů byla následně určena výtěžnost metody MEPS pro biologický vzorek (Obr. 28, Tab. 17). Pro všechny analyty byla získána výtěžnost v rozmezí 90 až 105 procent. Výjimkou byl o-hydroxyatorvastatin a atorvastatin lakton u vzorku číslo 1 a rosuvastatin lakton, jehož výtěžnost se pohybovala okolo 140%. I přesto, že výsledky byly velmi příznivé, bylo třeba ověřit přítomnost matricový efektů, které mohly výsledek ovlivnit jak negativně, tak pozitivně. Ověření těchto efektů bylo provedeno v následující validaci vyvinuté metody Průměr RV 103,8 96,4 103,3 101,2 RV-de 95,8 98,1 98,0 97,3 RVL 138,7 140,5 140,3 139,8 p-ohat 102,9 103,8 102,3 103,0 o-ohat 83,2 78,6 81,6 81,1 AT 97,8 101,5 100,3 99,9 ATL 84,7 106,7 95,0 95,4 Tab. 17: Výtěžnost optimalizované metody MEPS pro všechny analyty. 59
60 RV RV-de RVL p-ohat o-ohat AT ATL Obr. 28: Výtěžnost optimalizované metody MEPS pro všechny analyty. 60
61 5.5 Opakovatelnost a validace metody Opakovatelnost metody Po optimalizaci všech podmínek byla ověřena opakovatelnost metody na třech různých koncentračních hladinách v deseti po sobě následujících měřeních za stejných podmínek. Byl ověřen retenční čas a plocha píků všech analytů. SST byl proveden na třech hladinách 10, 100 a 500 nmol/l pro všechny stanovované látky. Opakovatelnost retenčních časů vyjádřená jako %RSD byla pro všechny analyty < 1% a opakovatelnost plochy píku byla < 5% (Tab. 18). ANALYTY RSD- t R [%] RSD plocha píku [%] L1 L2 L3 L1 L2 L3 RV 0,13 0,10 0,11 1,34 1,32 2,88 DES RV 0,25 0,31 0,46 3,29 1,84 2,11 RVL 0,13 0,01 0,11 3,21 2,89 3,96 p-oh-at 0,11 0,12 0,00 2,51 1,37 2,08 o-oh-at 0,20 0,24 0,07 2,19 1,40 2,41 AT 0,10 0,08 0,04 2,05 0,94 2,56 ATL 0,07 0,07 0,09 3,86 0,83 2,27 Tab. 18: Opakovatelnost metody. RSD (Relativní směrodatná odchylka, L1, L2, L3 koncentrační hladiny analytů 10, 100 a 500 nmol/l Validace metody V rámci validace metody byly ověřeny následující parametry metody: linearita, správnost, přesnost, matricové efekty, selektivita, limit detekce a limit kvantifikace. Metoda byla zvalidována v souladu s ICH směrnicí. Pro kvantitativní zhodnocení bylo využito vnitřních izotopicky značených standardů. Linearita Linearita metody byla ověřena na dvou typech kalibračních křivek. Nejprve byla ověřena linearita pro všechny analyty pomocí standardní kalibrační křivky v rozsahu 0, nmol/l, kde jako rozpouštědlo byla využita mobilní fáze odpovídající počátečnímu složení mobilní fáze. Následně byla také ověřena linearita pro matricovou 61
62 kalibrační křivku, kde matrici vzorku tvořilo sérum. Tato kalibrační křivka byla také proměřena v rozmezí 0, nmol/l pro všechny zjišťované analyty. K vyhodnocení linearity obou typů kalibračních křivek byl použit korelační koeficient (r 2 ), který dosahoval pro všechny analyty hodnot větších než 0,999. Výjimkou byl atorvastatin, pro který byla hodnota korelačního koeficientu rovna hodnotě 0,998 (Tab. 19). DES RV RV RVL p-oh AT o-oh AT AT ATL LINEARITA (r 2 ) matricová kalibrační křivka LINEARITA (r 2 ) - kalibrační křivka standardu 0,9991 0,9996 0,9990 0,9997 0,9993 0,9998 0,9982 0,9991 0,9998 0,9995 0,9991 0,9996 0,9998 0,9998 Tab. 19: Linearita - standardní a matricová kalibrační křivka pro všechny analyty. Analyt RV RVL RV-de Rovnice přímky 0,371552*x + 1,2541 0,87067*x + 2,4911 0, *x + 0,19923 Analyt AT ATL o-ohat p-ohat Rovnice přímky 1,04525 *x + 1, ,29473*x + 2, ,453153*x + 0,6407 0,431045*x + 1,30181 Tab. 20: Rovnice přímek standardů. Správnost a přesnost metody Správnost a přesnost metody byla ověřena na třech koncentračních hladinách ve třech provedeních. Koncentrační hladiny, které byly použity, byly 500, 50 a 5 nmol/l pro všechny analyty. Přesnost metody stanovená jako % RSD dosahovala hodnot nižších než 15 procent. Výtěžnost metody, která určuje správnost měření, se pohybovala v rozmezí 70 až 125 procenty pro všechny analyty (Tab. 21). 62
63 DES RV RV RVL p-oh AT o-oh AT AT ATL SPRÁVNOST METODY [ recovery %] PŘESNOST METODY [RSD %] L1 104,4 102,1 113,2 84,7 71,0 100,0 94,5 L2 99,9 102,5 119,6 85,5 77,0 103,3 92,9 L3 109,7 113,7 122,1 101,1 85,1 110,2 100,2 L1 1,9 0,6 0,5 1,4 3,4 1,4 2,7 L2 4,0 5,2 2,1 5,4 2,7 2,7 5,6 L3 3,3 7,3 14,8 3,6 12,5 6,6 6,9 Tab. 21: Správnost a přesnost metody pro všechny analyty stanovená na třech koncentračních hladinách (L1, L2, L3-500, 50 a 5 nmol/l). Matricové efekty K ověření matricových efektů byly použity dva různě připravené vzorky pro každý analyt, a to čisté sérum a blankové sérum upravené metodou MEPS a následně obohacené analyty. Pro vyhodnocení těchto efektů byla využita metoda postextrakčního přídavku (Obr. 18) i metoda postkolonového přídavku. Matricové efekty byly ověřeny na třech koncentračních hladinách - 500, 50 a 5 nmol/l. DES RV RV RVL p-oh AT o-oh AT AT ATL MATRICOVÉ EFEKTY [%] L1 96,0 100,4 98,8 98,0 99,4 102,3 99,3 L2 94,1 100,2 97,0 98,3 95,9 98,1 93,9 L3 105,1 99,4 99,6 89,7 95,7 98,3 89,9 Tab. 22: Ověření matricových efektů na třech koncentračních hladinách L1, L2, L3. Z výsledků plyne, že kvantitavní hodnocení analytů není matricovými efekty významně ovlivněno. Selektivita Selektivita byla ověřena proměřením lidského séra bez obsahu analytů, které bylo upraveno vyvinutou metodou MEPS. Bylo ověřeno, že v retenčních časech analyzovaných látek nejsou eluovány nečistoty a jiné balastní látky, které by analýzu mohly ovlivnit Obr
64 Obr. 29: Ověření selektivity nástřikem blanku. 64
65 Limit kvantifikace a limit detekce Limit detekce (LOD) a limit kvantifikace (LOQ) byly stanoveny pro vzorky, které byly připraveny za použití lidského séra, na základě matricové kalibrační křivky. Hodnoty LOD se pohybují v rozmezí do 1 nmol/l pro všechny analyty a LOQ v rozmezí 0,5 2,5 nmol/l pro všechny analyty, s tím, že mimo atoravastatin lakton a N- desmethylrosuvastatin nepřesahuje hodnota LOQ 1 nmol/l (Tab. 23). Metoda je tedy dostatečně citlivá a vhodná k použití pro monitorování lékových hladin rosuvastatinu, atorvastatinu a jejich metabolitů biologických vzorků pacientů. DES RV RV RVL p-oh AT o-oh AT AT ATL LOD [nmol/l] 0,75 0,3 0,3 0,3 0,15 0,15 0,75 LOQ [nmol/l] 2, ,5 1 2,5 Tab. 23: Limit detekce a kvantifikace pro všechny analyty. 65
66 6. Závěr Byla vyvinuta a optimalizována metoda pro analýzu biologického vzorku s obsahem rosuvastatinu, atorvastatinu a jejich metabolitů za použití izotopicky značených vnitřních standardů, kterými byly zvoleny deuteriem značený rosuvastatin a atorvastatin jako referenční látky pro každou skupinu analytů. Následně byla ověřena opakovatelnost metody byla stanovena relativní směrodatná odchylka pro retenční časy a plochy píků pro všechny analyty na třech koncentračních hladinách. Požadavek pro opakovatelnost retenčního času (hodnota nižší než 1%) byl splněn pro všechny analyty. Požadavek pro opakovatelnost ploch píků (maximálně 5-10%) byl také splněn pro všechny analyty, protože hodnoty RSD byly nižší než 5%. Byla vyvinuta a optimalizována metoda MEPS na standardním vzorku pro úpravu biologického vzorku. Nejtěžším krokem byla volba elučního činidla a sorbentu z důvodu rozdílných vlastností analytů. Bylo třeba vybrat nejvhodnější činidlo a sorbent tak, aby byla zaručena výtěžnost metody. Výsledný postup je uveden v Tab. 24. Objem činidla Použité činidlo Kondicionace kolonky 3x100 µl ACN Kondicionace kolonky 3x100 µl 0,1 M AmAc ph 4,5 Nanesení vzorku 50 µl Promytí 2x100 µl 0,1 M AmAc ph 4,5 Promytí 100 µl Promývací činidlo Eluce analytů 100 µl Eluční činidlo Promytí 100 µl Eluční činidlo Tab. 24: Výsledný postup metody MEPS. Promývací činidlo - ACN:0,01 M AmAc 4,5 ph 5:95; Eluční činidlo ACN:0,1 M AmAc 4,5 ph 95:5. Takto optimalizovaná metoda musela být následně aplikovaná na biologický materiál sérum, přičemž bylo třeba ověřit, zda výsledné hodnoty nejsou ovlivňovány interferencemi z matrice. Určení matricových efektů bylo zahrnuto do validace metody. Poté byla metoda zvalidována dle ICH podmínek. Ověřena byla linearita, správnost, přesnost, LOD, LOQ a matricové efekty (Tab. 25). Z výsledků je patrné, že metoda je lineární, vysoce citlivá s LOQ 0,5 2,5 nmol/l pro všechny analyty. 66
67 VALIDACE METODY DES p-oh RV RVL RV AT o-oh AT AT ATL LINEARITA (r 2 ) matricová kalibrační křivka 0,9991 0,9996 0,9990 0,9997 0,9993 0,9998 0,9982 LINEARITA (r 2 ) - kalibrační křivka standardu 0,9991 0,9998 0,9995 0,9991 0,9996 0,9998 0,9998 L1 104,4 102,1 113,2 84,7 71,0 100,0 94,5 SPRÁVNOST METODY L2 99,9 102,5 119,6 85,5 77,0 103,3 92,9 [% recovery] L3 109,7 113,7 122,1 101,1 85,1 110,2 100,2 L1 1,9 0,6 0,5 1,4 3,4 1,4 2,7 PŘESNOST METODY L2 4,0 5,2 2,1 5,4 2,7 2,7 5,6 [RSD %] L3 3,3 7,3 14,8 3,6 12,5 6,6 6,9 L1 96,0 100,4 98,8 98,0 99,4 102,3 99,3 MATRICOVÉ EFEKTY L2 94,1 100,2 97,0 98,3 95,9 98,1 93,9 [%] L3 105,1 99,4 99,6 89,7 95,7 98,3 89,9 LOD [nmol/l] 0,75 0,3 0,3 0,3 0,15 0,3 0,75 LOQ [nmol/l] 2, ,5 1 2,5 Tab. 25:Souhrn výsledků validace metody pro analýzu atorvastatinu a rosuvastatinu z biologického materiálu. L1, L2, L3 = koncentrace 500, 50 a 5 nmol/l, LOD, LOQ = hodnoty pro matricovou kalibrační křivku. Mezi výhody této metody patří především rychlost a poskytování správných a přesných výsledků. Propojení metody UHPLC-MS/MS s metodou MEPS navíc umožňuje rychlejší prostupnost vzorků laboratoří (není časově náročná na provedení), snižuje spotřebu používaných rozpouštědel a není invazivní metodou pro pacienty, protože k analýze postačí velmi malé množství biologického vzorku. V tomto případě je to 50 μl. Tato metoda však vyžaduje určitou zručnost a zkušenosti analytika. 67
68 7. Seznam použité literatury [1] L. Nováková, D. Šatínský, P. Solich, Trends Anal. Chem. 27 (2008) 352. [2] Hing-Biu Lee, T. E. Peart, M. Lewina Svoboda, S. Backus, Chemosphere 77 (2009) [3] H. Lüllman, K. Mohr, L. Hein, Barevný atlas farmakologie, 3. české vydání, Grada, [4] J. Hartl, M. Doležal, J. Křinková, M. Miletín, V. Opletalová, Farmaceutická chemie III (2004), 47. [5] H. Vaverková, Remedia 4 (2008), 324. [6] online [7] [8] J. M. McKenney, Clin. Cardiol. Vol. 26 (Suppl. III), III-32 III-38 (2003). [9] J. Vlček, D. Fialová a kol. Klinická farmacie I, 1. Vydání, Grada, [10] K. Górecká, I. Tilšer, P. Nachtigal, M. Kopecký, J. Vlček, Remedia 4 (2004). [11] S. L. Di Stasi, T. D. MacLeod, J. D. Winters, S. A. Binder-Macleod, Phys. Ther. 90 (2010) [12] S. S Tomlinson, K. K Mangione, Phys. Ther. 85 (2005) 459. [13] M.V.N. KumarTalluria,n, AnithaKalyankarb, SrinivasRagampeta, J. Pharm. Anal. 2 (2012) 285. [14] S. R. Polagani, N. R. Pilli, R. Gajula, V. Gandu. J. Pharm. Anal. (2012) v tisku. [15] Y. Shah, Z. Iqbal, L. Ahmad, A. Khan, M. I. Khan, S. Nazir, F. Nasir. J. Chromatogr. B 879 (2011) 557. [16] M.M.K. Sharaf El-Din, F. M.M. Salama, M. W.I. Nassar, K. A.M. Attia, M. M.Y. Kaddah. J. Pharm. Anal. 2 (2012) 200. [17] V. Sree Janardhanan, R. Manavalan, K. Valliappan. Arabian J. Chem. (2012) v tisku [18] R. Nirogi, K. Mudigonda, V. Kandikere, J. of Pharm. And Biomed. Anal. 44 (2007) 379. [19] jaa&url=http%3a%2f%2fwww.aapsj.org%2fabstracts%2fam_2008%2faaps pdf&ei=fwoqucslmypyswau7ic4cg&usg=afqjcngcrkf905u_p8aeiar60ieqpgk5 ia&sig2=dvzfqkszcrgqyc3pxfxuhw&bvm=bv ,d. online [20] T. R. Kumar, N. R. Shitut, P. K. Kumar, M. C. A. Vinu, V. Venkata, P. Kumar, R. Mullangi, N. R. Srinivas. Biomed. Chromatogr.20 (2006) 881. [21] R. V. S. Nirogi, V. N. Kandikere, M. Shukla, K. Mudigonda, S. Maurya, R. Boosi, Y.i Anjaneyulu. Biomed. Chromatogr. 20 (2006) 924. [22] R. K. Trivedi, R. R. Kallem, R. Mullangi, N. R. Srinivas. J. Pharm. Biomed. Anal. 39 (2009) 661. [23] K. A. Oudhoff, T. Sangster, E. Thomas, I. D. Wilson. J. Chromatogr.B 832 (2006)191. [24] S. S. Singh, K. Sharma, H. Patel, M. Jain, H. Shah, S. Gupta, P. Thakkar, N. Patel, S. P. Singh and B. B. Lohray. J. Braz. Chem. Soc.16 (2005) 944. [25] J. Gao, D. Zhong, X. Duan, X. Chen. J. Chromatogr. B 856 (2007) 35. [26] S. Vittal, N. R. Shitut, T. R. Kumar, M. C. A. Vinu, R. Mullangi, N. R. Srinivas. Biomed. Chromatogr. 20 (2006)1252. [27] H.-B. Lee, T.E. Peart, M. Lewina Svoboda, S. Backus. Chemosphere 77 (2009) [28] SNFXTs9NhHlJ2Kcd7H4z4Z7ZU2Ea3Zs8OIo0PwLxLW9LBOCz4woZEEZz06yOrau40SH- 68
69 LNCdEtpzIQg71H09kMM8GqPqVJb1aSRiXUt5pecIRvKnefk2dhSUPSo2qN&q=cache%3AQPi PCuWWYIcJ%3Awww.druginfo.com%2Fresource_library%2Fposters%2FRosuvastatin_ASMS_ 2008.pdf%20&docid=bc269f793af205eb8a45d2f209c0069a&a=bi&pagenumber=1&w=800. online [29] B. Di, M.-X. Su, F. Yu, L. J. Qu, L. P. Zhao, M.-Ch. Cheng, L.-P. He. J. Chromatogr. B 868 (2008) 95. [30] K. Lan, X. Jiang, Y. Li, L. Wang, J. Zhou, Q. Jiang, L. Ye. J. Pharm. Biomed. Anal. 44 (2007)540. [31] D.-H. Xu, Z.-R. Ruan, Q. Zhou, H. Yuan, B. Jiang. Rapid Commun. Mass Spectrom.20 (2006) [32] C. K. Hull, A. D. Penman, C. K. Smith, P.D. Martin. J. Chromatogr. 772 (2002) 219. [33] L.Nováková, H. Vlčková. Anal. Chim. Acta 656 (2009) 8. [34] L. Nováková, L. Matysová, P. Solich. Talanta 68 (2006) 908. [35] online [36] online online [37] P. Klouda, Moderní analytické metody. Ostrava, [38] online [39] online [40] J. Havliš. Základy hmotnostní spektrometrie, úvod, ionizační techniky. Přípravný kurz Základy hmotnostní spektrometrie 13. Ročník letní školy MS, Srní, [41] online [42] online [43] M. Himmelsbach. J. Chromatogr. B (2012) 3. [44] M. Gilar, E. S. P. Bouvier, B. J. Compton. J. Chromatogr. A 909 (2001)111. [45] L. Nováková. J. Chromatogr. A (2012) v tisku. [46] R.M. Smith. J. Chromatogr. A 1000 (2003) 3. [47] Y. Chen, Z. Guo, X. Wang, Ch. Qiu. J. Chromatogr. A 1184 (2008) 191. [48] online [49] online [50] online [51] online [52] S.K. Poole, C.F. Poole. J. Chromatogr. B 797 (2003) 3. [53] abtechold/soubory/operace/separacni_metody/extrakce.pdf+&hl=en&gl=cz&pid=bl&srcid=adgeesidi gu2clewgrwqfnwwvklsidypvredeblvzca1wuulydvig5yfikwzjdpgqtm3qxnovxmgyhf mgjqrnnuszfw2if8hrj_jzhdg9qsclm7jk7tcvd25fr_1bixm88_1skc17wo8&sig=ahietbti guui6yrykao5mjfbaullu5aaaa online
70 [54] on_and_isolation online [55] N6svgNMoG1_AUE_ZXRrC6Tp8pqRhjSLDPu3bOznebMyJX16mUJwRyespuZgJrfvvD8yzL34f u6vci53haiydrt07p13ldmqtxg4brggiyadufoyordeme2apyaw4as_ovs43m&sig=ahiet bqxnamb8xurzpcos69yxkv6rktlyq online [56] online [57] M-C. Hennion. J. Chromatogr. A 856 (1999) 3. [58] C. F. Poole. Trends in Anal. Chem. 22 (2003). [59] online [60] M. Hakkarainen. Polymer Degradation and Stability 95 (2010) 270. [61] A. Jain, K.K. Verma. Anal. Chim. Acta 706 (2011) 37. [62] A. Sarafraz-Yazdi, A. Amiri. Trends in Anal. Chem. 29 (2010). [63] M. I. Pinto, G. Sontag, R. J. Bernardino, J. P. Noronha. Microchem. J. 96 (2010) 225. [64] online [65] H. Vlčková, D. Solichová, M. Bláha, P. Solich, L. Nováková. J. Pharm. Biomed. Anal. 55 (2011) 301. [66] Z.i Altun, M. Abdel-Rehim. Anal. Chim. Acta 630 ( 2008 ) 116. [67] M. Abdel-Rehim. Anal. Chim. Acta 701 (2011) 119. [68] M. Abdel-Rehim. J. Chromatogr. A 1217 (2010) [69] Y. Chen, Z. Guo, X. Wang, Ch. Qiu. J. Chromatogr. A 1184 (2008) 191. [70] L. Junting, Ch. Peng, O. Suzuki. Forensic Science International 97 (1998) 93. [71] T. Kumazawa, X. P. Lee, K. Sato, O. Suzuki. Anal. Chim. Acta 492 (2003) 49. [72] H. Guan, W. E. Brewer, S. T. Garris, S. L. Morgan. J. Chromatogr. A 1217 (2010) [73] F. David, P. Sandra J. Chromatogr. A 1152 (2007) 54. [74] M. Kawaguchi, R. Ito, K. Saito, H. Nakazawa. J. Pharm. Biomed. Anal. 40 (2006) 500. [75] N. Delaunay-Bertoncini, M.-C. Hennion. J. Pharm. Biomed. Anal. 34 (2004) 717. [76] S. Tombelli, M. Minunni, M. Mascini. Biosensors and Bioelectronics 20 (2005) [77] M.-M. Zheng, S.-T. Wang, W.-K. Hu, Y.-Q. Feng. J. Chromatogr. A 1217 (2010) [78] Y. Gou, R. Eisert, J. Pawliszyn. J. Chromatogr. A 873 (2000) 137. [79] S. Souverain, S. Rudaz, J.-L Veuthey. Journal Chromatogr. B 801 (2004) 141. [80] W.M. Mullett.J. Biochem. Biophys. Methods 70 (2007) 263. [81] L. Ynddal, S.H. Hansen. J. Chromatogr. A 1020 (2003) 59. [82] J. Šabartová. Věstník SÚKL 1/1994. Str. 6. [83] Český lékopis Str [84] F.T. Peters, O. H. Drummer, F. Musshoff. Forensic Science International 165 (2007) 216. [85] L. Nováková. Matricové efekty v LC/MS analýze. 13. Ročník letní školy MS, Srní, [86] P.J. Taylor. Clinical Biochemistry 38 (2005) 328. [87] E. Klapková, R. Uřinovská, R. Průša. Klin. Biochem. Metab., 19 (2011) 5. 70
71 [88] F. Gosetti, E. Mazzucco, D. Zampieri, M. C. Gennaro. J. Chromatogr. A 1217 (2010) [89] P. Svoboda. Diplomová práce Vývoj UHPLC-MS/MS metody pro stanovení skupiny statinů a jejich metabolitů (2013). [90] V. Pilařová, H. Vlčková, P. Svoboda, L. Nováková. Chem. Listy 106 (2012) 96 [91] H. Vlčková et al. J. of Pharm. And Biomed. Anal. 55 (2011)
72 8. Přílohy 8.1 Abstrakty a publikace O cenu MERCK 2012, Praha 72

73 73
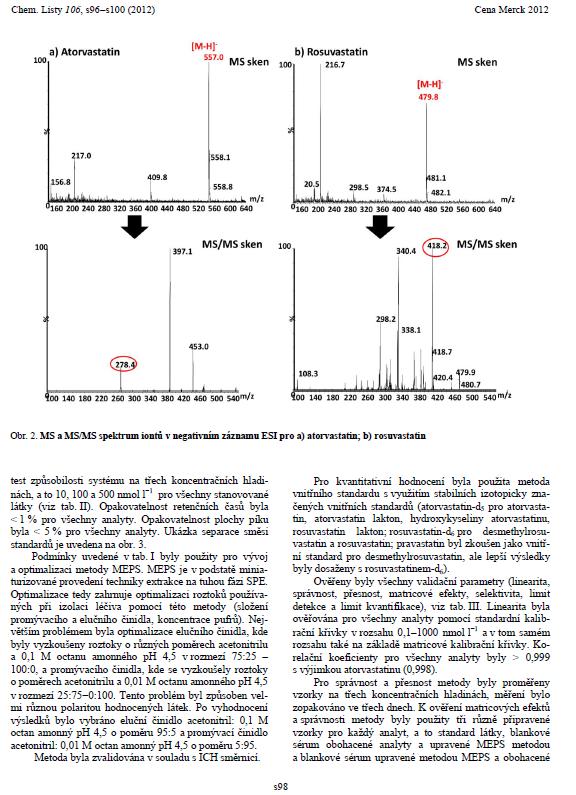
74 74
75 75
76 76
77 8.1.2 IMMS 2012, Olomouc Comparison of manual and semiautomatic approaches of MEPS as sample preparation technique in UHPLC-MS/MS methods for determination of statins in biological samples Vlčková H. 1, Pilařová V. 1, Svoboda P. 1, Nováková L. 1, Solich P. 1 1 Department of Analytical Chemistry, Faculty of Pharmacy in Hradec Králové, Charles University in Prague, Heyrovského 1203, Hradec Králové, Czech Republic hana.vlckova@faf.cuni.cz Biological samples are very complex matrices therefore the choice of suitable sample preparation technique before LC analysis is crucial. In contrast to fast LC, conventional sample preparation approaches are still high labor intensive and time-consuming. Microextraction by packed sorbent (MEPS) as a miniaturized SPE is logical extension of SPE for the analysis of biological fluids. MEPS is implemented by needle and syringe containing 1-2 mg of sorbent in the barrel of syringe. Sample preparation takes place on the packed bed. The sample can be drawn through the syringe manually or semi-automatically by automatic evol pipette. Two MEPS-UHPLC-MS/MS methods for determination of statins in biological samples were developed and validated. The first one for determination of atorvastatin, simvastatin and their derivates in human serum employs the manual approach of MEPS and the second one for the determination atorvastatin, rosuvastatin and their derivates employs the semi-automatic approach of MEPS. The analytical column Acquity BEH C18, mobile phase mixture of acetonitrile and 0.5mM ammonium acetate ph 4.0 and gradient elution were used. Electrospray ionisation (ESI) was selected as the suitable ionization technique. Individual parameters of ESI and triple quadrupole analyser were optimized. Quantification of all analytes was performed using SRM (selected reaction monitoring) transitions. For the both MEPS procedures C8 sorbent and 50µl of sample were used and 100µl of mixture of acetonitrile and 0.01M ammonium acetate ph 4.5 (95/5) was selected as the convenient elution solvent. Both developed methods were validated in terms of: linearity (>0.9990), accuracy ( %), precision (<7%) and matrix effect (<10%). Advantages and disadvantages of both MEPS-UHPLC- MS/MS methods (manual and semi-automatic approach MEPS) were compared. The advantages such as rapidity, simplicity and miniaturization, while maintaining sufficient selectivity, precision and accuracy, were directly confirmed for both MEPS methods. However during development of MEPS method using manual approach disadvantages including strong dependence of analyte recovery on continual movement of plunger and nonaccurate manual injection of very small volumes of sample (< 50 µl) were found. This is a critical feature of manual manipulation, which requires skilled operators. 77
78 Semi-automatic approach using evol hand- held automated analytical syringe which removes disadvantages of manual attitude was evaluated as a more suitable for the MEPS extraction and routine analysis of large number of biological samples. The study was supported by projects of Charles university in Prague UNCE 17/
79 79
80 8.1.3 Studentská vědecká konference , Farmaceutická fakulta Univerzity Karlovy v Hradci Králové Optimization of sample preparation step for UHPLC-MS/MS analysis of atorvastatin, rosuvastatin and their metabolites Pilařová V 1, Vlčková H 1, Svoboda P 1, Nováková L 1 1 Charles University in Prague, Faculty of Pharmacy in Hradec Králové, Department of Analytical Chemistry pilarovv@faf.cuni.cz Statins are very important group of drugs which are used to treat hypercholesterolemia and to prevent cardiovascular diseases. They are HMG-CoA reductase inhibitors. HMG-CoA reductase is the key enzyme that catalyzes the synthesis of cholesterol in human body. Statins exist in two forms acid and lactone form. Acid form is the effective form, while lactone form is transformed to open hydroxy-acid in human body. Both statins atorvastatin and rosuvastatin belong to the most frequently used statines because of their biological half-live and other pharmacological properties. Figure 30: Non-effective lactone form a) atorvastatin lactone c) rosuvastatin lactone; Effective open hydroxyacid form b) atorvastatin d) rosuvastatin. 80
81 A simple, sensitive and rapid ultra high performance liquid chromatography tandem mass spectometry (UHPLC-MS/MS) method was developed and validated for quantification of atorvastatin, rosuvastatin and their metabolites in human serum using atorvastatin-d 5 and rosuvastatin-d 6 as internal standards. MEPS (microextraction by packed sorbent) method was used for sample preparation of biological material. C8 was used as an extraction sorbent. Washing and elution reagents consisting of acetonitrile and ammonium acetate in various ratios were optimized. The mixture of acetonitrile and 0.1 M ammonium acetate ph 4.5 in a ratio of 95:5 was chosen as an elution agent. A mixture of acetonitrile and 0.01M ammonium acetate ph 4.5 in ratio of 5:95 was chosen as washing agent. The method was applied to the biological material (human serum). The analytes were separated using the Acquity BEH C18 column (50 x 2.1 mm, 1.7 µm, Waters) with gradient mobile phase (mixture of ammonium acetate and acetonitrile) and detected by electrospray (ESI) tandem mass spectrometry. Figure 31: SRM for standard analytes mixture of atorvastatin, atorvastatin lactone, p-hydroxyatorvastatin, o-hydroxyatorvastatin, atorvastatin-d5, rosuvastatin, rosuvastatin lactone, rosuvastatin-d6, N- desmethylrosuvastatin, pravastatin. Dissolved in ACN and 0.5 mm ammonium acetate ph 4.0 (30:70), Concentration of analytes was 5*10-7 mol/l. HPLC conditions: stationary phase Acquity BEH C18 (50 x
82 mm, 1.7 µm, Waters); mobile phase ACN and 0.5 mm ammonium acetate ph 4.0; ph 4.0; flow rate 0.3 ml/min; injection 5 μl; gradient elution. Finally the method was validated. The linearity, accuracy, precision, selectivity of the method were verified. The limit of detection and quantification and matrix effects were verified too. The method was linear with correlation coefficients in the range from to Limit of detection (LOD) ranged from 0.25 to 1.0 nmol / l and limit of quantification (LOQ) from 0.5 to 2.5 nmol / l. Precision was less than 15%, recovery, which indicates the accuracy, was in the range of 70 and 125% for all of analytes. The study was supported by SVV and UNCE /
83 8.1.4 Česká chromatografická škola HPLC 2013, Seč Optimization of sample preparation step for UHPLC-MS/MS analysis of atorvastatin, rosuvastatin and their metabolites Pilařová V 1, Vlčková H 1, Svoboda P 1, Nováková L 1 1 Charles University in Prague, Faculty of Pharmacy in Hradec Králové, Department of Analytical Chemistry, Heyrovského 1203, 50005, Hradec Králové pilarovaveronika@gmail.com Keywords: rosuvastatin, atorvastatin, MEPS, UHPLC-MS/MS. Rosuvastatin and atorvastatin are drugs used to treat hypercholesterolemia and to prevent cardiovascular diseases. They inhibit 3-hydroxy-3-methylglutaryl-Coenzym A (HMG- CoA) reductase which is the key enzyme that catalyzes the synthesis of cholesterol in human body. Both statins, rosuvastatin and atorvastatin, have advantageous pharmacological properties and long biological half-lives, therefore they are the most frequently used statins in clinical practice. Statins exists in two forms acid and lactone form. Acid form is the effective form, while lactone form is transformed to open hydroxy-acid in human body. Figure 1: Non-effective lactone form a) atorvastatin lactone c) rosuvastatin lactone; Effective open hydroxyacid form b) atorvastatin d) rosuvastatin. 83
84 Ultra high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS) method was developed and validated for quantification of atorvastatin, rosuvastatin and their metabolites (atorvastatin lactone, p- hydroxyatorvastatin, o-hydroxyatorvastatin, rosuvastatin lactone, N- desmethylrosuvastatin) in human serum using atorvastatin-d 5 and rosuvastatin-d 6 as internal standards. MEPS (microextraction by packed sorbent) method was used for sample preparation of biological material. The analytes were separated using the Acquity BEH C18 column (50 x 2.1 mm, 1.7 µm, Waters) with gradient mobile phase (mixture of 0.1 mm ammonium acetate ph 4.0 and acetonitrile) and detected by electrospray (ESI) tandem mass spectrometry. C8 was used as an extraction sorbent in MEPS procedure. Washing and elution reagents consisting of acetonitrile and ammonium acetate in various ratios were optimized. The mixture of acetonitrile and 0.1 M ammonium acetate ph 4.5 in a ratio of 95:5 was chosen as an elution agent. A mixture of acetonitrile and 0.01M ammonium acetate ph 4.5 in ratio of 5:95 was chosen as a washing agent. The method was applied to the biological material (human serum). Finally the method was validated. The linearity, limit of detection and quantification, accuracy, precision, selectivity of the method and matrix effects were verified. The method was linear with correlation coefficients in the range from to Limit of detection (LOD) ranged from 0.25 to 1.0 nmol/l and limit of quantification (LOQ) from 0.5 to 2.5 nmol/l. Method precision was good with RSD < 15%. Recovery, which indicates the method accuracy, was in the range of 70 and 125% for all of analytes. Analytes were not significantly influenced by matrix effects, which were verified at 3 concentration levels (500, 50, 5 mmol/l) by post-extraction addition method. This work was supported by the grant projects SVV and UNCE /
85 85
Klinická a farmaceutická analýza. Petr Kozlík Katedra analytické chemie
 Klinická a farmaceutická analýza Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz http://web.natur.cuni.cz/~kozlik/ 1 Spojení separačních technik s hmotnostní spektrometrem Separační
Klinická a farmaceutická analýza Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz http://web.natur.cuni.cz/~kozlik/ 1 Spojení separačních technik s hmotnostní spektrometrem Separační
HMOTNOSTNÍ SPEKTROMETRIE
 HMOTNOSTNÍ SPEKTROMETRIE MASS SPECTROMETRY (MS) Alternativní názvy (spojení s GC, LC, CZE, ITP): Hmotnostně spektrometrický (selektivní) detektor Mass spectrometric (selective) detector (MSD) Spektrometrie
HMOTNOSTNÍ SPEKTROMETRIE MASS SPECTROMETRY (MS) Alternativní názvy (spojení s GC, LC, CZE, ITP): Hmotnostně spektrometrický (selektivní) detektor Mass spectrometric (selective) detector (MSD) Spektrometrie
HMOTNOSTNÍ SPEKTROMETRIE - kvalitativní i kvantitativní detekce v GC a LC - pyrolýzní hmotnostní spektrometrie - analýza polutantů v životním
 HMOTNOSTNÍ SPEKTROMETRIE - kvalitativní i kvantitativní detekce v GC a LC - pyrolýzní hmotnostní spektrometrie - analýza polutantů v životním prostředí - farmakokinetické studie - kvantifikace proteinů
HMOTNOSTNÍ SPEKTROMETRIE - kvalitativní i kvantitativní detekce v GC a LC - pyrolýzní hmotnostní spektrometrie - analýza polutantů v životním prostředí - farmakokinetické studie - kvantifikace proteinů
Chromatografie. Petr Breinek
 Chromatografie Petr Breinek Chromatografie-I 2012 Společným znakem všech chromatografických metod je kontinuální dělení složek analyzované směsi mezi dvěma fázemi. Pohyblivá fáze (mobilní), eluent Nepohyblivá
Chromatografie Petr Breinek Chromatografie-I 2012 Společným znakem všech chromatografických metod je kontinuální dělení složek analyzované směsi mezi dvěma fázemi. Pohyblivá fáze (mobilní), eluent Nepohyblivá
MODERNÍ TRENDY V PŘÍPRAVĚ VZORKU K ANALÝZE S VYUŽITÍM INSTRUMENTÁLNÍCH TECHNIK
 MODERNÍ TRENDY V PŘÍPRAVĚ VZORKU K ANALÝZE S VYUŽITÍM INSTRUMENTÁLNÍCH TECHNIK Lucie Nováková Farmaceutická fakulta UK, Hradec Králové OUTLINE: 1) VÝBĚR METODY PRO PŘÍPRAVU VZORKU 2) STABILITA VZORKU 3)
MODERNÍ TRENDY V PŘÍPRAVĚ VZORKU K ANALÝZE S VYUŽITÍM INSTRUMENTÁLNÍCH TECHNIK Lucie Nováková Farmaceutická fakulta UK, Hradec Králové OUTLINE: 1) VÝBĚR METODY PRO PŘÍPRAVU VZORKU 2) STABILITA VZORKU 3)
SPE je metoda vhodná pro rychlou přípravu vzorků, která užívá
 Extrakce na pevné fázi (SPE) Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Extrakce na pevné fázi (SPE) (Solid Phase Extraction) SPE je metoda vhodná pro rychlou přípravu vzorků,
Extrakce na pevné fázi (SPE) Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Extrakce na pevné fázi (SPE) (Solid Phase Extraction) SPE je metoda vhodná pro rychlou přípravu vzorků,
Hmotnostní spektrometrie - Mass Spectrometry (MS)
") Hmotnostní spektrometrie - Mass Spectrometry (MS) Další pojem: Hmotnostně spektrometrický (selektivní) detektor - Mass spectrometric (selective) detector (MSD) Spektrometrie - metoda založená na interakci
Hmotnostní spektrometrie - Mass Spectrometry (MS) Další pojem: Hmotnostně spektrometrický (selektivní) detektor - Mass spectrometric (selective) detector (MSD) Spektrometrie - metoda založená na interakci
Analytická technika HPLC-MS/MS a možnosti jejího využití v hygieně
 Analytická technika HPLC-MS/MS a možnosti jejího využití v hygieně Šárka Dušková 24. září 2015-61. konzultační den Hodnocení expozice chemickým látkám na pracovištích 1 HPLC-MS/MS HPLC high-performance
Analytická technika HPLC-MS/MS a možnosti jejího využití v hygieně Šárka Dušková 24. září 2015-61. konzultační den Hodnocení expozice chemickým látkám na pracovištích 1 HPLC-MS/MS HPLC high-performance
Hmotnostní spektrometrie. Historie MS. Schéma MS
 Hmotnostní spektrometrie MS mass spectrometry MS je analytická technika, která se používá k měření poměru hmotnosti ku náboji (m/z) u iontů původně studium izotopového složení dnes dynamicky se vyvíjející
Hmotnostní spektrometrie MS mass spectrometry MS je analytická technika, která se používá k měření poměru hmotnosti ku náboji (m/z) u iontů původně studium izotopového složení dnes dynamicky se vyvíjející
Vysokoúčinná kapalinová chromatografie
 Vysokoúčinná kapalinová chromatografie HPLC High Performance Liquid Chromatography Vysokoúčinná...X... Vysoceúčinná kapalinová chromatografie RRLC Rapid Resolution Liquid Chromatography Rychle rozlišovací
Vysokoúčinná kapalinová chromatografie HPLC High Performance Liquid Chromatography Vysokoúčinná...X... Vysoceúčinná kapalinová chromatografie RRLC Rapid Resolution Liquid Chromatography Rychle rozlišovací
Vývoj MEPS metody pro UHPLC-MS/MS stanovení entekaviru v ultrafiltrátu ledvin
 Univerzita Karlova v Praze Farmaceutická fakulta v Hradci Králové Katedra analytické chemie Vývoj MEPS metody pro UHPLC-MS/MS stanovení entekaviru v ultrafiltrátu ledvin Vedoucí diplomové práce: RNDr.
Univerzita Karlova v Praze Farmaceutická fakulta v Hradci Králové Katedra analytické chemie Vývoj MEPS metody pro UHPLC-MS/MS stanovení entekaviru v ultrafiltrátu ledvin Vedoucí diplomové práce: RNDr.
EXTRAKČNÍ METODY. Studijní materiál. 1. Obecná charakteristika extrakce. 2. Extrakce kapalina/kapalina LLE. 3. Alkalická hydrolýza
 Studijní materiál EXTRAKČNÍ METODY 1. Obecná charakteristika extrakce 2. Extrakce kapalina/kapalina LLE 3. Alkalická hydrolýza 4. Soxhletova extrakce 5. Extrakce za zvýšené teploty a tlaku PLE, ASE, PSE
Studijní materiál EXTRAKČNÍ METODY 1. Obecná charakteristika extrakce 2. Extrakce kapalina/kapalina LLE 3. Alkalická hydrolýza 4. Soxhletova extrakce 5. Extrakce za zvýšené teploty a tlaku PLE, ASE, PSE
Hmotnostní spektrometrie
 Hmotnostní spektrometrie Princip: 1. Ze vzorku jsou tvořeny ionty na úrovni molekul, nebo jejich zlomků (fragmentů), nebo až volných atomů dodáváním energie, např. uvolnění atomů ze vzorku nebo přímo rozštěpení
Hmotnostní spektrometrie Princip: 1. Ze vzorku jsou tvořeny ionty na úrovni molekul, nebo jejich zlomků (fragmentů), nebo až volných atomů dodáváním energie, např. uvolnění atomů ze vzorku nebo přímo rozštěpení
LABORATOŘ OBORU I ÚSTAV ORGANICKÉ TECHNOLOGIE (111) Použití GC-MS spektrometrie
 Použití GC-MS spektrometrie") LABORATOŘ OBORU I ÚSTAV ORGANICKÉ TECHNOLOGIE (111) C Použití GC-MS spektrometrie Vedoucí práce: Doc. Ing. Petr Kačer, Ph.D., Ing. Kamila Syslová Umístění práce: laboratoř 79 Použití GC-MS spektrometrie
LABORATOŘ OBORU I ÚSTAV ORGANICKÉ TECHNOLOGIE (111) C Použití GC-MS spektrometrie Vedoucí práce: Doc. Ing. Petr Kačer, Ph.D., Ing. Kamila Syslová Umístění práce: laboratoř 79 Použití GC-MS spektrometrie
UNIVERZITA KARLOVA V PRAZE FARMACEUTICKÁ FAKULTA V HRADCI KRÁLOVÉ Katedra analytické chemie
 UNIVERZITA KARLOVA V PRAZE FARMACEUTICKÁ FAKULTA V HRADCI KRÁLOVÉ Katedra analytické chemie Vývoj UHPLC-MS/MS metody pro stanovení pravastatinu, rosuvastatinu a jejich metabolitů Vedoucí diplomové práce:
UNIVERZITA KARLOVA V PRAZE FARMACEUTICKÁ FAKULTA V HRADCI KRÁLOVÉ Katedra analytické chemie Vývoj UHPLC-MS/MS metody pro stanovení pravastatinu, rosuvastatinu a jejich metabolitů Vedoucí diplomové práce:
HMOTNOSTNÍ SPEKTROMETRIE
 HMOTNOSTNÍ SPEKTROMETRIE A MOŽNOSTI JEJÍHO SPOJENÍ SE SEPARAČNÍMI METODAMI SEPARACE chromatografie CGC, GC x GC HPLC, UPLC, UHPLC, CHIP-LC elektromigrační m. CZE, CITP INTERFACE SPOJENÍ x ROZHRANÍ GC vyhřívaná
HMOTNOSTNÍ SPEKTROMETRIE A MOŽNOSTI JEJÍHO SPOJENÍ SE SEPARAČNÍMI METODAMI SEPARACE chromatografie CGC, GC x GC HPLC, UPLC, UHPLC, CHIP-LC elektromigrační m. CZE, CITP INTERFACE SPOJENÍ x ROZHRANÍ GC vyhřívaná
jako markeru oxidativního
 Monitoring koncentrace 8-isoprostanu jako markeru oxidativního stresu v kondenzátu vydechovaného vzduchu Lukáš Chytil Ústav organické technologie Úvod Cíl: - nalezení vhodného analytické metody pro analýzu
Monitoring koncentrace 8-isoprostanu jako markeru oxidativního stresu v kondenzátu vydechovaného vzduchu Lukáš Chytil Ústav organické technologie Úvod Cíl: - nalezení vhodného analytické metody pro analýzu
Vysokoúčinná kapalinová chromatografie. Petr Kozlík Katedra analytické chemie
 Vysokoúčinná kapalinová chromatografie Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz 1 Sylabus přednášky: Praxe v HPLC Mobilní fáze Chromatografická kolona Spoje v HPLC Vývoj chromatografické
Vysokoúčinná kapalinová chromatografie Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz 1 Sylabus přednášky: Praxe v HPLC Mobilní fáze Chromatografická kolona Spoje v HPLC Vývoj chromatografické
No. 1- určete MW, vysvětlení izotopů
 No. 1- určete MW, vysvětlení izotopů ESI/APCI + 325 () 102 (35) 327 (33) 326 (15) 328 (5) 150 200 250 300 350 400 450 500 ESI/APCI - 323 () 97 (51) 325 (32) 324 (13) 326 (6) 150 200 250 300 350 400 450
No. 1- určete MW, vysvětlení izotopů ESI/APCI + 325 () 102 (35) 327 (33) 326 (15) 328 (5) 150 200 250 300 350 400 450 500 ESI/APCI - 323 () 97 (51) 325 (32) 324 (13) 326 (6) 150 200 250 300 350 400 450
ÚSTAV CHEMIE A ANALÝZY POTRAVIN
 VYSOKÁ ŠKOLA CHEMICKO-TECHNOLOGICKÁ V PRAZE ÚSTAV CHEMIE A ANALÝZY POTRAVIN Technická 5, 166 28 Praha 6 tel./fax.: + 420 220 443 185; jana.hajslova@vscht.cz LABORATOŘ Z ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ
VYSOKÁ ŠKOLA CHEMICKO-TECHNOLOGICKÁ V PRAZE ÚSTAV CHEMIE A ANALÝZY POTRAVIN Technická 5, 166 28 Praha 6 tel./fax.: + 420 220 443 185; jana.hajslova@vscht.cz LABORATOŘ Z ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU NEPOVOLENÝCH DOPLŇKOVÝCH LÁTEK METODOU LC-MS
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU NEPOVOLENÝCH DOPLŇKOVÝCH LÁTEK METODOU LC-MS 1 Účel a rozsah Tato metoda specifikuje podmínky pro stanovení nepovolených doplňkových látek Zn-bacitracinu,
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU NEPOVOLENÝCH DOPLŇKOVÝCH LÁTEK METODOU LC-MS 1 Účel a rozsah Tato metoda specifikuje podmínky pro stanovení nepovolených doplňkových látek Zn-bacitracinu,
Diagnostika bronchiálního. ho astmatu HPLC/MS analýzou. Kamila Syslová Ústav organické technologie
 Diagnostika bronchiálního ho astmatu HPLC/MS analýzou Kamila Syslová Ústav organické technologie Bronchiální astma Civilizační onemocnění rostoucí počet případů snižující se věková hranice prvních projevů
Diagnostika bronchiálního ho astmatu HPLC/MS analýzou Kamila Syslová Ústav organické technologie Bronchiální astma Civilizační onemocnění rostoucí počet případů snižující se věková hranice prvních projevů
INTERPRETACE HMOTNOSTNÍCH SPEKTER
 INTERPRETACE HMOTNOSTNÍCH SPEKTER Hmotnostní spektrometrie hmotnostní spektrometrie = fyzikálně chemická metoda založená na rozdělení hmotnosti iontů v plynné fázi podle jejich poměru hmotnosti a náboje
INTERPRETACE HMOTNOSTNÍCH SPEKTER Hmotnostní spektrometrie hmotnostní spektrometrie = fyzikálně chemická metoda založená na rozdělení hmotnosti iontů v plynné fázi podle jejich poměru hmotnosti a náboje
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU MELAMINU A KYSELINY KYANUROVÉ METODOU LC-MS
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU MELAMINU A KYSELINY KYANUROVÉ METODOU LC-MS 1 Rozsah a účel Postup je určen pro stanovení obsahu melaminu a kyseliny kyanurové v krmivech. 2 Princip
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU MELAMINU A KYSELINY KYANUROVÉ METODOU LC-MS 1 Rozsah a účel Postup je určen pro stanovení obsahu melaminu a kyseliny kyanurové v krmivech. 2 Princip
Iontové zdroje II. Iontový zdroj. Data. Vzorek. Hmotnostní analyzátor. Zdroj vakua. Iontové zdroje pracující za sníženého tlaku
 Iontové zdroje II. Iontové zdroje pracující za sníženého tlaku Elektronová/chemická ionizace Iontové zdroje pro spojení s planárními separacemi Ionizace laserem za účasti matrice Ambientní ionizační techniky
Iontové zdroje II. Iontové zdroje pracující za sníženého tlaku Elektronová/chemická ionizace Iontové zdroje pro spojení s planárními separacemi Ionizace laserem za účasti matrice Ambientní ionizační techniky
Hmotnostní detekce v separačních metodách
 Hmotnostní detekce v separačních metodách MC230P83 2/1 Z+Zk 4 kredity doc. RNDr. Josef Cvačka, Ph.D. Mgr. Martin Hubálek, Ph.D. Ústav organické chemie a biochemie AVČR, v.v.i. Flemingovo nám. 2, 166 10
Hmotnostní detekce v separačních metodách MC230P83 2/1 Z+Zk 4 kredity doc. RNDr. Josef Cvačka, Ph.D. Mgr. Martin Hubálek, Ph.D. Ústav organické chemie a biochemie AVČR, v.v.i. Flemingovo nám. 2, 166 10
Inovace bakalářského a navazujícího magisterského studijního programu v oboru Bezpečnost a kvalita potravin (reg. č. CZ.1.07/2.2.00/28.
 Inovace bakalářského a navazujícího magisterského studijního programu v oboru Bezpečnost a kvalita potravin (reg. č. CZ.1.07/2.2.00/28.0287) EXTRAKČNÍ METODY Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana
Inovace bakalářského a navazujícího magisterského studijního programu v oboru Bezpečnost a kvalita potravin (reg. č. CZ.1.07/2.2.00/28.0287) EXTRAKČNÍ METODY Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana
Chromatografie. Petr Breinek. Chromatografie_2011 1
 Chromatografie Petr Breinek Chromatografie_2011 1 Společným znakem všech chromatografických metod je kontinuální rozdělování složek analyzované směsi vzorku mezi dvěma fázemi. Nepohyblivá fáze (stacionární
Chromatografie Petr Breinek Chromatografie_2011 1 Společným znakem všech chromatografických metod je kontinuální rozdělování složek analyzované směsi vzorku mezi dvěma fázemi. Nepohyblivá fáze (stacionární
HPLC/MS tělních tekutin nový rozměr v medicinální diagnostice
 HPLC/MS tělních tekutin nový rozměr v medicinální diagnostice Lukáš Chytil Ústav organické technologie VŠCHT Praha Medicinální diagnostika a hmotnostní spektrometrie Medicinální diagnostika: - Klasické
HPLC/MS tělních tekutin nový rozměr v medicinální diagnostice Lukáš Chytil Ústav organické technologie VŠCHT Praha Medicinální diagnostika a hmotnostní spektrometrie Medicinální diagnostika: - Klasické
Vývoj postupu extrakce pro izolaci pravastatinu a jeho metabolitu z biologického materiálu pomocí SPE
 Univerzita Karlova v Praze Farmaceutická fakulta v Hradci Králové Katedra analytické chemie Vývoj postupu extrakce pro izolaci pravastatinu a jeho metabolitu z biologického materiálu pomocí SPE RIGORÓZNÍ
Univerzita Karlova v Praze Farmaceutická fakulta v Hradci Králové Katedra analytické chemie Vývoj postupu extrakce pro izolaci pravastatinu a jeho metabolitu z biologického materiálu pomocí SPE RIGORÓZNÍ
EXTRAKČNÍ METODY používané pro stanovení lipofilních a hydrofilních látek. Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana Borkovcová, Ph.D.
 EXTRAKČNÍ METODY používané pro stanovení lipofilních a hydrofilních látek Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana Borkovcová, Ph.D. EXTRAKČNÍ METODY Úvod rozdělení látek podle polarity extrakce lipofilních
EXTRAKČNÍ METODY používané pro stanovení lipofilních a hydrofilních látek Mgr. Romana Kostrhounová, Ph. D. RNDr. Ivana Borkovcová, Ph.D. EXTRAKČNÍ METODY Úvod rozdělení látek podle polarity extrakce lipofilních
P. Martinková, R. Jobánek, D. Pospíchalová. Stanovení vybraných léčiv v čistírenském kalu
 P. Martinková, R. Jobánek, D. Pospíchalová Stanovení vybraných léčiv v čistírenském kalu PPCP Pharmaceutical and Personal Care Products (farmaka a produkty osobní potřeby) Do životního prostředí se dostávají
P. Martinková, R. Jobánek, D. Pospíchalová Stanovení vybraných léčiv v čistírenském kalu PPCP Pharmaceutical and Personal Care Products (farmaka a produkty osobní potřeby) Do životního prostředí se dostávají
Separační metody v analytické chemii. Kapalinová chromatografie (LC) - princip
 - princip") Kapalinová chromatografie (LC) - princip Kapalinová chromatografie (Liquid chromatography, zkratka LC) je typ separační metody, založené na rozdílné distribuci dělených látek ve směsi mezi dvě různé nemísitelné
Kapalinová chromatografie (LC) - princip Kapalinová chromatografie (Liquid chromatography, zkratka LC) je typ separační metody, založené na rozdílné distribuci dělených látek ve směsi mezi dvě různé nemísitelné
ROLE SEPARAČNÍCH METOD
 ROLE SEPARAČNÍCH METOD Redukce nežádoucích složek - ruší analýzu, poškozují přístroj Rozdělení - frakcionace vzorku podle zvolené charakteristiky Cílená analýza - vysoce selektivní postup Necílená analýza
ROLE SEPARAČNÍCH METOD Redukce nežádoucích složek - ruší analýzu, poškozují přístroj Rozdělení - frakcionace vzorku podle zvolené charakteristiky Cílená analýza - vysoce selektivní postup Necílená analýza
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ
 LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ STANOVENÍ BIOLOGICKY AKTIVNÍCH LÁTEK POMOCÍ VYSOKOÚČINNÉ CHROMATOGRAFIE VE SPOJENÍ S HMOTNOSTNÍ SPEKTROMETRIÍ (LC-MS) Garant úlohy: Ing. Vojtěch Hrbek 1
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ STANOVENÍ BIOLOGICKY AKTIVNÍCH LÁTEK POMOCÍ VYSOKOÚČINNÉ CHROMATOGRAFIE VE SPOJENÍ S HMOTNOSTNÍ SPEKTROMETRIÍ (LC-MS) Garant úlohy: Ing. Vojtěch Hrbek 1
Studijní materiál. Úvod do problematiky extrakčních metod. Vypracoval: RNDr. Ivana Borkovcová, Ph.D.
 Studijní materiál Úvod do problematiky extrakčních metod Vypracoval: RNDr. Ivana Borkovcová, Ph.D. Úvod do problematiky extrakčních metod Definice, co je to extrakce separační proces v kontaktu jsou dvě
Studijní materiál Úvod do problematiky extrakčních metod Vypracoval: RNDr. Ivana Borkovcová, Ph.D. Úvod do problematiky extrakčních metod Definice, co je to extrakce separační proces v kontaktu jsou dvě
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ. Stanovení těkavých látek
 LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení těkavých látek (metoda: plynová chromatografie s hmotnostně spektrometrickým detektorem) Garant úlohy: doc. Ing. Jana Pulkrabová, Ph.D. 1 OBSAH
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení těkavých látek (metoda: plynová chromatografie s hmotnostně spektrometrickým detektorem) Garant úlohy: doc. Ing. Jana Pulkrabová, Ph.D. 1 OBSAH
Separační metody v analytické chemii. Plynová chromatografie (GC) - princip
 - princip") Plynová chromatografie (GC) - princip Plynová chromatografie (Gas chromatography, zkratka GC) je typ separační metody, kdy se od sebe oddělují složky obsažené ve vzorku a které mohou být převedeny do plynné
Plynová chromatografie (GC) - princip Plynová chromatografie (Gas chromatography, zkratka GC) je typ separační metody, kdy se od sebe oddělují složky obsažené ve vzorku a které mohou být převedeny do plynné
Jednotné pracovní postupy zkoušení krmiv Vydání 1 STANOVENÍ OBSAHU KOKCIDIOSTATIK METODOU LC-MS
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU KOKCIDIOSTATIK METODOU LC-MS 1 Účel a rozsah Postup specifikuje podmínky pro stanovení diclazurilu, halofuginonu, lasalocidu, maduramicinu, monensinu,
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU KOKCIDIOSTATIK METODOU LC-MS 1 Účel a rozsah Postup specifikuje podmínky pro stanovení diclazurilu, halofuginonu, lasalocidu, maduramicinu, monensinu,
Jednotné pracovní postupy zkoušení krmiv Vydání 1 STANOVENÍ OBSAHU KOKCIDIOSTATIK METODOU LC-MS
 Strana 1 STANOVENÍ OBSAHU KOKCIDIOSTATIK METODOU LC-MS 1 Účel a rozsah Postup specifikuje podmínky pro stanovení diclazurilu, halofuginonu, lasalocidu, maduramicinu, monensinu, narasinu, nikarbazinu, robenidinu,
Strana 1 STANOVENÍ OBSAHU KOKCIDIOSTATIK METODOU LC-MS 1 Účel a rozsah Postup specifikuje podmínky pro stanovení diclazurilu, halofuginonu, lasalocidu, maduramicinu, monensinu, narasinu, nikarbazinu, robenidinu,
Spojení hmotové spektrometrie se separačními metodami
 Spojení hmotové spektrometrie se separačními metodami RNDr. Radomír Čabala, Dr. Katedra analytické chemie Přírodovědecká fakulta Univerzita Karlova Praha Spojení hmotové spektrometrie se separačními metodami
Spojení hmotové spektrometrie se separačními metodami RNDr. Radomír Čabala, Dr. Katedra analytické chemie Přírodovědecká fakulta Univerzita Karlova Praha Spojení hmotové spektrometrie se separačními metodami
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU MYKOTOXINŮ METODOU LC-MS - FUMONISIN B 1 A B 2
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU MYKOTOXINŮ METODOU LC-MS - FUMONISIN B 1 A B 2 1 Rozsah a účel Metoda je vhodná pro stanovení fumonisinů B 1 a B 2 v krmivech. 2 Princip Fumonisiny
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU MYKOTOXINŮ METODOU LC-MS - FUMONISIN B 1 A B 2 1 Rozsah a účel Metoda je vhodná pro stanovení fumonisinů B 1 a B 2 v krmivech. 2 Princip Fumonisiny
ULTRA PERFORMANCE LIQUID CHROMATOGRAPHY (UPLC) ULTRA-HIGH PERFORMANCE LIQUID CHROMATOGRAPHY (UHPC)
 ULTRA-HIGH PERFORMANCE LIQUID CHROMATOGRAPHY (UHPC)") ULTRA PERFORMANCE LIQUID CHROMATOGRAPHY (UPLC) ULTRA-HIGH PERFORMANCE LIQUID CHROMATOGRAPHY (UHPC) Pokroky v moderních separačních metodách, 2012 Eva Háková CHARAKTERISTIKA UPLC Nová, velmi účinná separační
ULTRA PERFORMANCE LIQUID CHROMATOGRAPHY (UPLC) ULTRA-HIGH PERFORMANCE LIQUID CHROMATOGRAPHY (UHPC) Pokroky v moderních separačních metodách, 2012 Eva Háková CHARAKTERISTIKA UPLC Nová, velmi účinná separační
Gelová permeační chromatografie
 Gelová permeační chromatografie (Gel Permeation Chromatography - GPC) - separační a čisticí metoda - umožňuje separaci skupin sloučenin s podobnou molekulovou hmotností (frakcionace) - analyty jsou po
Gelová permeační chromatografie (Gel Permeation Chromatography - GPC) - separační a čisticí metoda - umožňuje separaci skupin sloučenin s podobnou molekulovou hmotností (frakcionace) - analyty jsou po
Úvod do strukturní analýzy farmaceutických látek
 Úvod do strukturní analýzy farmaceutických látek Garant předmětu: doc. Ing. Bohumil Dolenský, Ph.D. A28, linka 4110, dolenskb@vscht.cz Hmotnostní spektrometrie II. Příprava předmětu byla podpořena projektem
Úvod do strukturní analýzy farmaceutických látek Garant předmětu: doc. Ing. Bohumil Dolenský, Ph.D. A28, linka 4110, dolenskb@vscht.cz Hmotnostní spektrometrie II. Příprava předmětu byla podpořena projektem
Vysokoúčinná kapalinová chromatografie. Petr Kozlík Katedra analytické chemie
 Vysokoúčinná kapalinová chromatografie Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz http://web.natur.cuni.cz/~kozlik/ 1 Vysokoúčinná kapalinová chromatografie Teorie HPLC Praktické
Vysokoúčinná kapalinová chromatografie Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz http://web.natur.cuni.cz/~kozlik/ 1 Vysokoúčinná kapalinová chromatografie Teorie HPLC Praktické
Chromatofokusace. separace proteinů na základě jejich pi vysoké rozlišení. není potřeba připravovat ph gradient zaostřovací efekt jednoduchost
 Chromatofokusace separace proteinů na základě jejich pi vysoké rozlišení není potřeba připravovat ph gradient zaostřovací efekt jednoduchost Polypufry - amfolyty Stacionární fáze Polybuffer 96 - ph 9-6
Chromatofokusace separace proteinů na základě jejich pi vysoké rozlišení není potřeba připravovat ph gradient zaostřovací efekt jednoduchost Polypufry - amfolyty Stacionární fáze Polybuffer 96 - ph 9-6
UNIVERZITA PARDUBICE Fakulta chemicko-technologická Katedra analytické chemie. Nám. Čs. Legií 565, Pardubice.
 UNIVERZITA PARDUBICE Fakulta chemicko-technologická Katedra analytické chemie Nám. Čs. Legií 565, 532 10 Pardubice 15. licenční studium INTERAKTIVNÍ STATISTICKÁ ANALÝZA DAT Semestrální práce VYUŽITÍ TABULKOVÉHO
UNIVERZITA PARDUBICE Fakulta chemicko-technologická Katedra analytické chemie Nám. Čs. Legií 565, 532 10 Pardubice 15. licenční studium INTERAKTIVNÍ STATISTICKÁ ANALÝZA DAT Semestrální práce VYUŽITÍ TABULKOVÉHO
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU MYKOTOXINŮ METODOU LC-MS - aflatoxin B1, B2, G1 a G2
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU MYKOTOXINŮ METODOU LC-MS - aflatoxin B1, B2, G1 a G2 1 Rozsah a účel Metoda je vhodná pro stanovení aflatoxinů B1, B2, G1 a G2 v krmivech. 2 Princip
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU MYKOTOXINŮ METODOU LC-MS - aflatoxin B1, B2, G1 a G2 1 Rozsah a účel Metoda je vhodná pro stanovení aflatoxinů B1, B2, G1 a G2 v krmivech. 2 Princip
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ. Stanovení těkavých látek
 LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení těkavých látek (metoda: plynová chromatografie s hmotnostně spektrometrickým detektorem) Garant úlohy: Ing. Jaromír Hradecký, Ph.D. 1 OBSAH Základní
LABORATOŘ ANALÝZY POTRAVIN A PŘÍRODNÍCH PRODUKTŮ Stanovení těkavých látek (metoda: plynová chromatografie s hmotnostně spektrometrickým detektorem) Garant úlohy: Ing. Jaromír Hradecký, Ph.D. 1 OBSAH Základní
Příprava materiálu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253
 Příprava materiálu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Část 16 Iontová chromatografie Iontová chromatografie je speciální technika vyvinutá pro separaci anorganických iontů a organických
Příprava materiálu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Část 16 Iontová chromatografie Iontová chromatografie je speciální technika vyvinutá pro separaci anorganických iontů a organických
Stanovení esterů steroidů v krevním séru
 Stanovení esterů steroidů v krevním séru Ústav pro státní kontrolu veterinárních biopreparátů a léčiv (ÚSKVBL) Brno, Hudcova 56a Mgr. Martina Rejtharová Ing. Katarína Čačková rejtharova@uskvbl.cz cackova@uskvbl.cz
Stanovení esterů steroidů v krevním séru Ústav pro státní kontrolu veterinárních biopreparátů a léčiv (ÚSKVBL) Brno, Hudcova 56a Mgr. Martina Rejtharová Ing. Katarína Čačková rejtharova@uskvbl.cz cackova@uskvbl.cz
Analýza kofeinu v kávě pomocí kapalinové chromatografie
 Analýza kofeinu v kávě pomocí kapalinové chromatografie Kofein (obr.1) se jako přírodní alkaloid vyskytuje v mnoha rostlinách (např. fazolích, kakaových bobech, černém čaji apod.) avšak nejvíce je spojován
Analýza kofeinu v kávě pomocí kapalinové chromatografie Kofein (obr.1) se jako přírodní alkaloid vyskytuje v mnoha rostlinách (např. fazolích, kakaových bobech, černém čaji apod.) avšak nejvíce je spojován
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU DEKOCHINÁTU METODOU HPLC
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU DEKOCHINÁTU METODOU HPLC 1 Rozsah a účel Tato metoda specifikuje podmínky pro stanovení dekochinátu metodou vysokoúčinné kapalinové chromatografie
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU DEKOCHINÁTU METODOU HPLC 1 Rozsah a účel Tato metoda specifikuje podmínky pro stanovení dekochinátu metodou vysokoúčinné kapalinové chromatografie
Trendy v moderní HPLC
 Trendy v moderní HPLC Josef Cvačka, 5.1.2011 CHROMATOGRAFIE NA ČIPECH Miniaturizace separačních systémů Mikrofluidní čipy Mikrofabrikace Chromatografické mikrofluidní čipy s MS detekcí Praktické využití
Trendy v moderní HPLC Josef Cvačka, 5.1.2011 CHROMATOGRAFIE NA ČIPECH Miniaturizace separačních systémů Mikrofluidní čipy Mikrofabrikace Chromatografické mikrofluidní čipy s MS detekcí Praktické využití
Mobilní fáze. HPLC mobilní fáze 1
 Mobilní fáze 1 VLIV CHROMATOGRAFICKÝCH PODMÍNEK NA ELUČNÍ CHARAKTERISTIKY SEPAROVANÝCH LÁTEK - SLOŽENÍ MOBILNÍ FÁZE Složení mobilní fáze má vliv na eluční charakteristiky : účinnost kolony; kapacitní poměr;
Mobilní fáze 1 VLIV CHROMATOGRAFICKÝCH PODMÍNEK NA ELUČNÍ CHARAKTERISTIKY SEPAROVANÝCH LÁTEK - SLOŽENÍ MOBILNÍ FÁZE Složení mobilní fáze má vliv na eluční charakteristiky : účinnost kolony; kapacitní poměr;
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU REZIDUÍ POLÁRNÍCH PESTICIDŮ METODOU LC-MS
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU REZIDUÍ POLÁRNÍCH PESTICIDŮ METODOU LC-MS 1 Rozsah a účel Postup je určen pro analýzu reziduí účinných látek přípravků na ochranu rostlin v obilovinách,
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU REZIDUÍ POLÁRNÍCH PESTICIDŮ METODOU LC-MS 1 Rozsah a účel Postup je určen pro analýzu reziduí účinných látek přípravků na ochranu rostlin v obilovinách,
Zdroje iont používané v hmotnostní spektrometrii. Miloslav Šanda
 Zdroje iont používané v hmotnostní spektrometrii Miloslav Šanda Ionizace v MS Hmotnostní spektrometrie je fyzikáln chemická metoda, pi které se provádí separace iont podle jejich hmotnosti a náboje m/z
Zdroje iont používané v hmotnostní spektrometrii Miloslav Šanda Ionizace v MS Hmotnostní spektrometrie je fyzikáln chemická metoda, pi které se provádí separace iont podle jejich hmotnosti a náboje m/z
Stanovení sacharidů ve vybraných přírodních matricích pomocí kapalinové chromatografie s odpařovacím detektorem rozptylu světla (HPLC-ELSD)
") Stanovení sacharidů ve vybraných přírodních matricích pomocí kapalinové chromatografie s odpařovacím detektorem rozptylu světla (HPLC-ELSD) A) Ultrazvuková extrakce Ultrazvuková extrakce je významnou extrakční
Stanovení sacharidů ve vybraných přírodních matricích pomocí kapalinové chromatografie s odpařovacím detektorem rozptylu světla (HPLC-ELSD) A) Ultrazvuková extrakce Ultrazvuková extrakce je významnou extrakční
L 54/116 CS Úřední věstník Evropské unie
 L 54/116 CS Úřední věstník Evropské unie 26.2.2009 8. Výsledky kruhových testů V rámci ES byly provedeny kruhové testy, při nichž až 13 laboratoří zkoušelo čtyři vzorky krmiva pro selata, včetně jednoho
L 54/116 CS Úřední věstník Evropské unie 26.2.2009 8. Výsledky kruhových testů V rámci ES byly provedeny kruhové testy, při nichž až 13 laboratoří zkoušelo čtyři vzorky krmiva pro selata, včetně jednoho
Superkritická fluidní extrakce (SFE) Superkritická fluidní extrakce
 Superkritická fluidní extrakce") Superkritická fluidní extrakce (zkráceně SFE, z angl. Supercritical Fluid Extraction) = extrakce, kde extrakčním činidlem je tekutina v superkritickém stavu, tzv. superkritická (nadkritická) tekutina (zkráceně
Superkritická fluidní extrakce (zkráceně SFE, z angl. Supercritical Fluid Extraction) = extrakce, kde extrakčním činidlem je tekutina v superkritickém stavu, tzv. superkritická (nadkritická) tekutina (zkráceně
P. Martinková, D. Pospíchalová, R. Jobánek, M. Jokešová. Stanovení perfluorovaných organických látek v elektroodpadech
 P. Martinková, D. Pospíchalová, R. Jobánek, M. Jokešová Stanovení perfluorovaných organických látek v elektroodpadech Perfluorované a polyfluorované uhlovodíky (PFC,PFAS) Perfluorované - všechny vodíky
P. Martinková, D. Pospíchalová, R. Jobánek, M. Jokešová Stanovení perfluorovaných organických látek v elektroodpadech Perfluorované a polyfluorované uhlovodíky (PFC,PFAS) Perfluorované - všechny vodíky
Stanovení cholesterolu ve vaječném žloutku a mléce kapilární elektroforézou
 Stanovení cholesterolu ve vaječném žloutku a mléce kapilární elektroforézou Úkol Stanovte obsah cholesterolu ve vaječném žloutku a mléce pomocí kapilární elektroforézy. Teoretická část Cholesterol je steroidní
Stanovení cholesterolu ve vaječném žloutku a mléce kapilární elektroforézou Úkol Stanovte obsah cholesterolu ve vaječném žloutku a mléce pomocí kapilární elektroforézy. Teoretická část Cholesterol je steroidní
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU VITAMÍNU D METODOU LC/MS
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU VITAMÍNU D METODOU LC/MS 1 Účel a rozsah Tento postup specifikuje podmínky pro stanovení vitamínu D3 v krmivech metodou LC/MS. 2 Princip Zkušební
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU VITAMÍNU D METODOU LC/MS 1 Účel a rozsah Tento postup specifikuje podmínky pro stanovení vitamínu D3 v krmivech metodou LC/MS. 2 Princip Zkušební
DETEKTORY pro kapalinovou chromatografii. Izolační a separační metody, 2018
 DETEKTORY pro kapalinovou chromatografii Izolační a separační metody, 2018 Detektory v kapalinové chromatografii Typ detektoru Zkratka Měřená veličina Refraktometrický detektor RID index lomu Spektrofotometrický
DETEKTORY pro kapalinovou chromatografii Izolační a separační metody, 2018 Detektory v kapalinové chromatografii Typ detektoru Zkratka Měřená veličina Refraktometrický detektor RID index lomu Spektrofotometrický
Aplikační rozsah chromatografie
 Chromatografické metody II. Aplikační rozsah chromatografie Chromatografie Kapalinová chromatografie rozdělení Nízkotlaká (atmosferický tlak) LPC Střednětlaká (4 Mpa) FPLC Vysokotlaká (40 Mpa) HPLC Ultravysokotlaká
Chromatografické metody II. Aplikační rozsah chromatografie Chromatografie Kapalinová chromatografie rozdělení Nízkotlaká (atmosferický tlak) LPC Střednětlaká (4 Mpa) FPLC Vysokotlaká (40 Mpa) HPLC Ultravysokotlaká
 Vysokoúčinná kapalinová chromatografie High-Performance Liquid Chromatography (HPLC) Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Kapalinová chromatografie (LC) 1.1. Teorie kapalinové
Vysokoúčinná kapalinová chromatografie High-Performance Liquid Chromatography (HPLC) Příprava předmětu byla podpořena projektem OPPA č. CZ.2.17/3.1.00/33253 Kapalinová chromatografie (LC) 1.1. Teorie kapalinové
Klinická a farmaceutická analýza. Petr Kozlík Katedra analytické chemie
 Klinická a farmaceutická analýza Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz http://web.natur.cuni.cz/~kozlik/ 1 Sylabus přednášky: Validace Validační parametry Validace bioanalytické
Klinická a farmaceutická analýza Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz http://web.natur.cuni.cz/~kozlik/ 1 Sylabus přednášky: Validace Validační parametry Validace bioanalytické
Iontové zdroje II. Iontový zdroj. Data. Vzorek. Hmotnostní analyzátor. Zdroj vakua. Iontové zdroje pracující za sníženého tlaku
 MC230P43 Hmotnostní detekce v separačních metodách, 2015 Iontové zdroje II. Iontové zdroje pracující za sníženého tlaku Elektronová/chemická ionizace Iontové zdroje pro spojení s planárními separacemi
MC230P43 Hmotnostní detekce v separačních metodách, 2015 Iontové zdroje II. Iontové zdroje pracující za sníženého tlaku Elektronová/chemická ionizace Iontové zdroje pro spojení s planárními separacemi
3/7/2014. Dávkování vzorku LC/MS. Dávkování vzorku LC/MS
 motnostní spektrometrie (3) Josef Chudoba LC/MS BSA - dávkování vzorku u LC/MS - PLC chromatografie chromatogram, základní pojmy režimy PLC, mobilní fáze, kolony (stacionární fáze) - elektrosprejová ionizace
motnostní spektrometrie (3) Josef Chudoba LC/MS BSA - dávkování vzorku u LC/MS - PLC chromatografie chromatogram, základní pojmy režimy PLC, mobilní fáze, kolony (stacionární fáze) - elektrosprejová ionizace
Název: Vypracovala: Datum: 7. 2. 2014. Zuzana Lacková
 Název: Vypracovala: Zuzana Lacková Datum: 7. 2. 2014 Reg.č.projektu: CZ.1.07/2.4.00/31.0023 Název projektu: Partnerská síť centra excelentního bionanotechnologického výzkumu MĚLI BYCHOM ZNÁT: informace,
Název: Vypracovala: Zuzana Lacková Datum: 7. 2. 2014 Reg.č.projektu: CZ.1.07/2.4.00/31.0023 Název projektu: Partnerská síť centra excelentního bionanotechnologického výzkumu MĚLI BYCHOM ZNÁT: informace,
Stanovení fenolických látek pomocí kapalinové chromatografie
 Stanovení fenolických látek pomocí kapalinové chromatografie A) Princip extrakce podle Randalla Extrakci provádíme ve třech krocích: 1. Vaření V první fázi je extrakční prst obsahující vzorek ponořen do
Stanovení fenolických látek pomocí kapalinové chromatografie A) Princip extrakce podle Randalla Extrakci provádíme ve třech krocích: 1. Vaření V první fázi je extrakční prst obsahující vzorek ponořen do
mobilní fáze pohyblivá - obsahuje dělené látky, které mají různou afinitu ke stacionární fázi.
 separační metody Chromatografické metody Distribuce látky mezi dvě fáze: stacionární fáze nepohyblivá - ukotvený materiál mobilní fáze pohyblivá - obsahuje dělené látky, které mají různou afinitu ke stacionární
separační metody Chromatografické metody Distribuce látky mezi dvě fáze: stacionární fáze nepohyblivá - ukotvený materiál mobilní fáze pohyblivá - obsahuje dělené látky, které mají různou afinitu ke stacionární
Vysokoúčinná kapalinová chromatografie. Petr Kozlík Katedra analytické chemie
 Vysokoúčinná kapalinová chromatografie Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz 1 Aplikace HPLC Analýza složek životního prostředí Toxikologie Potravinářská analýza Farmaceutická
Vysokoúčinná kapalinová chromatografie Petr Kozlík Katedra analytické chemie e-mail: kozlik@natur.cuni.cz 1 Aplikace HPLC Analýza složek životního prostředí Toxikologie Potravinářská analýza Farmaceutická
Víme, co vám nabízíme
 PDF vygenerováno: 30.12.2016 5:20: Katalog / Laboratorní pomůcky / ace / Nástavce a filtrační špičky na injekční stříkačky Nástavec filtrační na injekční stříkačky MACHEREY-NAGEL Jednoúčelové nástavce
PDF vygenerováno: 30.12.2016 5:20: Katalog / Laboratorní pomůcky / ace / Nástavce a filtrační špičky na injekční stříkačky Nástavec filtrační na injekční stříkačky MACHEREY-NAGEL Jednoúčelové nástavce
Repetitorium chemie IX (2016) (teorie a praxe chromatografie)
 (teorie a praxe chromatografie)") Repetitorium chemie IX (2016) (teorie a praxe chromatografie) Chromatografie Podstatou je rozdělování složek směsi dávkovaného vzorku mezi dvěma fázemi Stacionární fáze je nepohyblivá (silikagel, celulóza,
Repetitorium chemie IX (2016) (teorie a praxe chromatografie) Chromatografie Podstatou je rozdělování složek směsi dávkovaného vzorku mezi dvěma fázemi Stacionární fáze je nepohyblivá (silikagel, celulóza,
VYUŽITÍ BEZKONTAKTNÍ VODIVOSTNÍ DETEKCE PRO HPLC SEPARACI POLYKARBOXYLÁTOVÝCH DERIVÁTŮ CYKLENU. Anna Hamplová
 VYUŽITÍ BEZKOTAKTÍ VODIVOSTÍ DETEKCE PRO HPLC SEPARACI POLYKARBOXYLÁTOVÝCH DERIVÁTŮ CYKLEU Anna Hamplová Univerzita Karlova v Praze, Přírodovědecká fakulta, Katedra analytické chemie Albertov 6, 128 43
VYUŽITÍ BEZKOTAKTÍ VODIVOSTÍ DETEKCE PRO HPLC SEPARACI POLYKARBOXYLÁTOVÝCH DERIVÁTŮ CYKLEU Anna Hamplová Univerzita Karlova v Praze, Přírodovědecká fakulta, Katedra analytické chemie Albertov 6, 128 43
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU REZIDUÍ POLÁRNÍCH PESTICIDŮ METODOU LC-MS
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU REZIDUÍ POLÁRNÍCH PESTICIDŮ METODOU LC-MS 1 Rozsah a účel Postup je určen pro analýzu reziduí účinných látek přípravků na ochranu rostlin v obilovinách,
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU REZIDUÍ POLÁRNÍCH PESTICIDŮ METODOU LC-MS 1 Rozsah a účel Postup je určen pro analýzu reziduí účinných látek přípravků na ochranu rostlin v obilovinách,
Repetitorium chemie IV (2014)
") Repetitorium chemie IV (2014) Chromatografie Podstatou je rozdělování složek směsi dávkovaného vzorku mezi dvěma fázemi Stacionární fáze je nepohyblivá (silikagel, celulóza, polymerní částice) Mobilní
Repetitorium chemie IV (2014) Chromatografie Podstatou je rozdělování složek směsi dávkovaného vzorku mezi dvěma fázemi Stacionární fáze je nepohyblivá (silikagel, celulóza, polymerní částice) Mobilní
Průtokové metody (Kontinuální měření v proudu kapaliny)
") Průtokové metody (Kontinuální měření v proudu kapaliny) 1. Přímé měření: analyzovaná kapalina většinou odvětvena + vhodný detektor 2. Kapalinová chromatografie (HPLC) Stanovení po předchozí separaci 3.
Průtokové metody (Kontinuální měření v proudu kapaliny) 1. Přímé měření: analyzovaná kapalina většinou odvětvena + vhodný detektor 2. Kapalinová chromatografie (HPLC) Stanovení po předchozí separaci 3.
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU MADURAMICINU A SEMDURAMICINU METODOU HPLC
 Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU MADURAMICINU A SEMDURAMICINU METODOU HPLC 1 Rozsah a účel Metoda specifikuje podmínky pro stanovení maduramicinu a semduramicinu v krmivech a premixech.
Národní referenční laboratoř Strana 1 STANOVENÍ OBSAHU MADURAMICINU A SEMDURAMICINU METODOU HPLC 1 Rozsah a účel Metoda specifikuje podmínky pro stanovení maduramicinu a semduramicinu v krmivech a premixech.
Detekce a detektory část 2
 Detekce a detektory část 2 Ivan Mikšík Fyziologický ústav AV ČR, v.v.i. Praha Spojení (spřažení) hmotnostní spektrometrie a separačních technik Analýza složitých směsí (nejdříve separace, poté analýza)
Detekce a detektory část 2 Ivan Mikšík Fyziologický ústav AV ČR, v.v.i. Praha Spojení (spřažení) hmotnostní spektrometrie a separačních technik Analýza složitých směsí (nejdříve separace, poté analýza)
Principy chromatografie v analýze potravin
 Principy chromatografie v analýze potravin živočišného původu p Ivana Borkovcová Ústav hygieny a technologie mléka FVHE VFU Brno, borkovcovai@vfu.cz Úvod, základní pojmy chromatografické systémy dělení
Principy chromatografie v analýze potravin živočišného původu p Ivana Borkovcová Ústav hygieny a technologie mléka FVHE VFU Brno, borkovcovai@vfu.cz Úvod, základní pojmy chromatografické systémy dělení
CS Úřední věstník Evropské unie L 54/85
 26.2.2009 CS Úřední věstník Evropské unie L 54/85 F. STANOVENÍ DICLAZURILU 2,6-dichlor-alfa-(4-chlorofenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3-H)yl)benzenacetonitril 1. Účel a rozsah Tato metoda
26.2.2009 CS Úřední věstník Evropské unie L 54/85 F. STANOVENÍ DICLAZURILU 2,6-dichlor-alfa-(4-chlorofenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3-H)yl)benzenacetonitril 1. Účel a rozsah Tato metoda
Úvod k biochemickému praktiku. Pavel Jirásek
 Úvod k biochemickému praktiku Pavel Jirásek Úvodní informace 4 praktika B1 B2 B3 B4 4 týdny 8 pracovních stolů rozdělení kruhu do 8 pracovních skupin (v každé 2-3 studenti) Co s sebou na praktika plášť
Úvod k biochemickému praktiku Pavel Jirásek Úvodní informace 4 praktika B1 B2 B3 B4 4 týdny 8 pracovních stolů rozdělení kruhu do 8 pracovních skupin (v každé 2-3 studenti) Co s sebou na praktika plášť
ADSORPČNÍ CHROMATOGRAFIE (LSC)
") EXTRAKCE TUHOU FÁZÍ ADSORPČNÍ CHROMATOGRAFIE (LSC) -rozdělení směsi látek (primární extrakt) na sloupci sorbentu ve skleněné koloně s fritou (cca 50 cm x 1 cm) -obvykle jde o selektivní adsorpci nežádoucích
EXTRAKCE TUHOU FÁZÍ ADSORPČNÍ CHROMATOGRAFIE (LSC) -rozdělení směsi látek (primární extrakt) na sloupci sorbentu ve skleněné koloně s fritou (cca 50 cm x 1 cm) -obvykle jde o selektivní adsorpci nežádoucích
Přímá analýza reálných vzorků hmotnostní spektrometrií s využitím nanodesorpčního elektrospreje (nano-desi-ms)
") Přímá analýza reálných vzorků hmotnostní spektrometrií s využitím nanodesorpčního elektrospreje (nano-desi-ms) Teorie: Desorpční elektrosprej (DESI) byl popsán v roce 2004 Zoltánem Takátsem. Jedná se o
Přímá analýza reálných vzorků hmotnostní spektrometrií s využitím nanodesorpčního elektrospreje (nano-desi-ms) Teorie: Desorpční elektrosprej (DESI) byl popsán v roce 2004 Zoltánem Takátsem. Jedná se o
Indentifikace molekul a kvantitativní analýza pomocí MS
 Indentifikace molekul a kvantitativní analýza pomocí MS Identifikace molekul snaha určit molekulovou hmotnost, sumární složení, strukturní části molekuly (funkční skupiny, aromatická jádra, alifatické
Indentifikace molekul a kvantitativní analýza pomocí MS Identifikace molekul snaha určit molekulovou hmotnost, sumární složení, strukturní části molekuly (funkční skupiny, aromatická jádra, alifatické
Jednotné pracovní postupy zkoušení krmiv STANOVENÍ OBSAHU SEMDURAMICINU METODOU HPLC
 Strana 1 STANOVENÍ OBSAHU SEMDURAMICINU METODOU HPLC 1 Rozsah a účel Postup specifikuje podmínky pro stanovení obsahu semduramicinu v krmivech metodou vysokoúčinné kapalinové chromatografie (HPLC) v koncentračním
Strana 1 STANOVENÍ OBSAHU SEMDURAMICINU METODOU HPLC 1 Rozsah a účel Postup specifikuje podmínky pro stanovení obsahu semduramicinu v krmivech metodou vysokoúčinné kapalinové chromatografie (HPLC) v koncentračním
Laboratoř ze speciální analýzy potravin II. Úloha 3 - Plynová chromatografie (GC-MS)
") 1 Úvod... 1 2 Cíle úlohy... 2 3 Předpokládané znalosti... 2 4 Autotest základních znalostí... 2 5 Základy práce se systémem GC-MS (EI)... 3 5.1 Parametry plynového chromatografu... 3 5.2 Základní charakteristiky
1 Úvod... 1 2 Cíle úlohy... 2 3 Předpokládané znalosti... 2 4 Autotest základních znalostí... 2 5 Základy práce se systémem GC-MS (EI)... 3 5.1 Parametry plynového chromatografu... 3 5.2 Základní charakteristiky
Zajištění správnosti výsledků analýzy kotininu a kreatininu
 Zajištění správnosti výsledků analýzy kotininu a kreatininu Š.Dušková, I.Šperlingová, L. Dabrowská, M. Tvrdíková, M. Šubrtová duskova@szu.cz sperling@szu.cz Oddělení pro hodnocení expozice chemickým látkám
Zajištění správnosti výsledků analýzy kotininu a kreatininu Š.Dušková, I.Šperlingová, L. Dabrowská, M. Tvrdíková, M. Šubrtová duskova@szu.cz sperling@szu.cz Oddělení pro hodnocení expozice chemickým látkám
METODY ČIŠTĚNÍ ORGANICKÝCH LÁTEK
 METODY ČIŠTĚNÍ ORGANICKÝCH LÁTEK Chemické sloučeniny se připravují z jiných chemických sloučenin. Tento děj se nazývá chemická reakce, kdy z výchozích látek (reaktantů) vznikají nové látky (produkty).
METODY ČIŠTĚNÍ ORGANICKÝCH LÁTEK Chemické sloučeniny se připravují z jiných chemických sloučenin. Tento děj se nazývá chemická reakce, kdy z výchozích látek (reaktantů) vznikají nové látky (produkty).
CRH/NPU I - Systém pro ultraúčinnou kapalinovou chromatografii (UHPLC) ve spojení s tandemovým hmotnostním spektrometrem (MS/MS)
 ve spojení s tandemovým hmotnostním spektrometrem (MS/MS)") ODŮVODNĚNÍ VEŘEJNÉ ZAKÁZKY v souladu s 156 zákona č. 137/2006, Sb., o veřejných zakázkách, ve znění pozdějších předpisů Nadlimitní veřejná zakázka na dodávky zadávaná v otevřeném řízení v souladu s ust.
ODŮVODNĚNÍ VEŘEJNÉ ZAKÁZKY v souladu s 156 zákona č. 137/2006, Sb., o veřejných zakázkách, ve znění pozdějších předpisů Nadlimitní veřejná zakázka na dodávky zadávaná v otevřeném řízení v souladu s ust.
Univerzita Karlova v Praze Farmaceutická Fakulta v Hradci Králové Katedra analytické chemie
 Univerzita Karlova v Praze Farmaceutická Fakulta v Hradci Králové Katedra analytické chemie Využití HPLC pro stanovení barviv ilegálně používaných v potravinách (diplomová práce) Vedoucí diplomové práce:
Univerzita Karlova v Praze Farmaceutická Fakulta v Hradci Králové Katedra analytické chemie Využití HPLC pro stanovení barviv ilegálně používaných v potravinách (diplomová práce) Vedoucí diplomové práce:
Hmotnostní spektrometrie
 Hmotnostní spektrometrie Podstatou hmotnostní spektrometrie je studium iontů v plynném stavu. Tato metoda v sobě zahrnuje tři hlavní části:! generování iontů sledovaných atomů nebo molekul! separace iontů
Hmotnostní spektrometrie Podstatou hmotnostní spektrometrie je studium iontů v plynném stavu. Tato metoda v sobě zahrnuje tři hlavní části:! generování iontů sledovaných atomů nebo molekul! separace iontů
Kapalinová chromatografie ve spojení s hmotnostní detekcí ( LC-MS )
") Úloha do laboratorního cvičení - Speciální metody Kapalinová chromatografie ve spojení s hmotnostní detekcí ( LC-MS ) Analýza bílého vína: stanovení organických kyselin Teoretická část úlohy: Chemické
Úloha do laboratorního cvičení - Speciální metody Kapalinová chromatografie ve spojení s hmotnostní detekcí ( LC-MS ) Analýza bílého vína: stanovení organických kyselin Teoretická část úlohy: Chemické