SEQUENCE ALIGNMENT MOLEKULÁRNÍ TAXONOMIE
|
|
|
- Bohumila Pešková
- před 6 lety
- Počet zobrazení:
Transkript
1 SEQUENCE ALIGNMENT gi gi Eat1 SpEat1 CG7206 DrEat1 C1orf26 46 LQELDNLKKGKML LHV-RQKAI 46 LQELDYLKSGKLS SKV-EDKAR 47 IQELDGLKKSPDIARDNDDTTN----QEHDRTI-GTLAR 49 LQELDGLKSESS STC-GYLAR 49 IKELDKLKSKYQS DCLQRVIAM 49 LQELDYLKSGKLS SKV-EDKAR 49 MQELDRMKEGKLL KRA-QHKAI MOLEKULÁRNÍ TAXONOMIE
2 OSNOVA co to je sequence alignment? jak se dělá sequence alignment? jaké použít programy?
 se srovnávají tzv. alignmentem HTTP://LGIMAGES.S3.AMAZONAWS.")
3 REKONSTRUKCE EVOLUČNÍ HISTORIE rekonstrukce na základě srovnávání znaků v molekulární taxonomii se používají sekvence sekvence (DNA, RNA, proteiny) se srovnávají tzv. alignmentem
4 CO JE SEQUENCE ALIGNMENT? v biologii - seřazení sekvencí DNA, RNA nebo proteinů pod sebe tak, že (dle evoluce) odpovídající si amimokyseliny leží pod sebou (při zachování jejich pořadí) cílem je odhalit oblasti podobnosti mezi sekvencemi, které poukazuji na funkční, strukturní nebo evoluční podobnost v informatice - série operací, kterými se změní jedna sekvence v druhou (substituce, inzerce, delece)
5 PROČ SROVNÁVAT SEKVENCE? informace o funkci informace o struktuře proteinu informace o evoluci, nalézání příbuzenských vztahů odvodit, které aminokyseliny si odpovidají (homologie)
6 CO JE SEQUENCE ALIGNMENT? >Q61287 Q61287_MOUSE ALPHA-GLOBIN - MUS MUSCULUS (MOUSE). MVLSGEDKSNIKAAWGKIGGHGAEYVAEALERMFASFPTTKTYFPHFDVSHGSAQVKGHG KKVADALASAAGHLDDLPGALSALSDLHAHKLRVDPVNFKLLSHCLLVTLASHHPADFTP AVHASLDKFLASVSTVLTSKYR >Q5XMD6 Q5XMD6_9AVES ALPHA A HEMOGLOBIN - OXYURA MACCOA. MVLSAADKTNVKGVFSKIGGHADDYGAETLERMFVAYPQTKTYFPHFDLQHGSAQIKAHG KKVAAALVEAVNHIDDISGALSKLSDLHAQKLRVDPVNFKFLGHCFLVVVAIHHPSALTP EVHASLDKFMCAVGAVLTAKYR Q61287 Q61287_MOUSE MVLSGEDKSNIKAAWGKIGGHGAEYVAEALERMFASFPTTKTYFPHFDVSHGSAQVKGHG 60 Q5XMD6 Q5XMD6_9AVES MVLSAADKTNVKGVFSKIGGHADDYGAETLERMFVAYPQTKTYFPHFDLQHGSAQIKAHG 60 ****. **:*:*..:.*****. :* **:*****.::* *********:.*****:*.** Q61287 Q61287_MOUSE KKVADALASAAGHLDDLPGALSALSDLHAHKLRVDPVNFKLLSHCLLVTLASHHPADFTP 120 Q5XMD6 Q5XMD6_9AVES KKVAAALVEAVNHIDDISGALSKLSDLHAQKLRVDPVNFKFLGHCFLVVVAIHHPSALTP 120 **** **..*..*:**:.**** ******:**********:*.**:**.:* ***: :** Q61287 Q61287_MOUSE AVHASLDKFLASVSTVLTSKYR 142 Q5XMD6 Q5XMD6_9AVES EVHASLDKFMCAVGAVLTAKYR 142 ********:.:*.:***:***

7 PROČ JSOU SI SEKVENCE PODOBNÉ? náhodou analogické homologické
8 POKUD JSOU HOMOLOGICKÉ... SEQUENCE 1 VLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKF-DRF-KHLKTEAEMKAS.=ALI =ID SEQUENCE 2 VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPH-FD--L----SHG-S -- SEQUENCE 1 EDLKKHGVTVLTALGAILKKKGHHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSR.=ALI =ID SEQUENCE 2 AQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAH -- SEQUENCE 1 HPGDFGADAQGAMNKALELFRKDIAAKYKELGY.=ALI =ID SEQUENCE 2 LPAEFTPAVHASLDKFLASVSTVLTSKY---R- lze usuzovat, že: shoda = obě aminokyseliny zůstaly zachovány ve stejné formě v jaké se nacházejí u nejbližšího společného předka neshoda = mutace (substituce) jedné z aminokyselin po odštěpení ze společného předka mezera = inzerce nebo delece v jedné ze sekvencí po odštěpení od společného předka
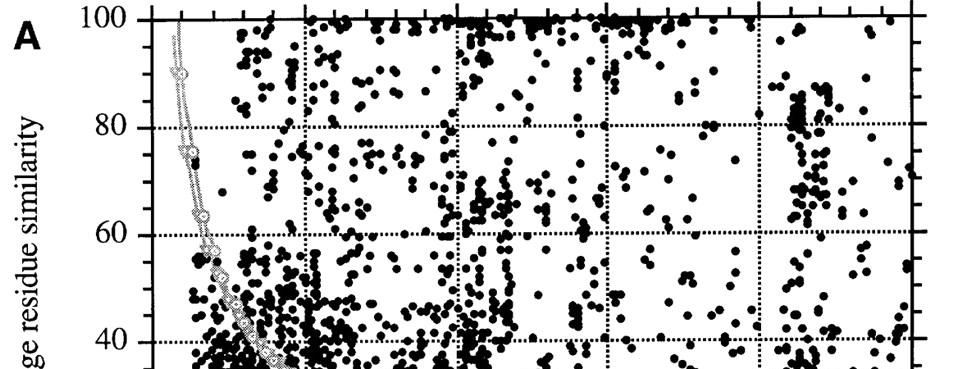


9 SEQUENCE IDENTITA/HOMOLOGIE HOMOLOGNÍ PROTEINY ROST, 1999
10 SEQUENCE IDENTITA/HOMOLOGIE sekvenční identita > 35% - pravděpodobně homolog sekvenční identita = 20-35% ( twilight zone ; Doolittle) - může být homolog sekvenční identita < 20% - midnight zone (Rost) - sekvence zcela nedostatečná k určení homologie

11 PROTEIN NEBO DNA? při určování homologie se obvykle používá proteinový alignment 20 aminokyselin versus 4 nucleotidy => mnohem větší pravděpodobnost shody na každé pozici v DNA, proteinový alignment proto informativnější na druhou stranu - genetický kód je degenerovaný => různé kodony často kódují stejnou aminokyselinu - rozdíl v DNA alignmentu se vůbec neprojeví v proteinovém alignmentu a neovlivní vlastnosti proteinu (genového produktu) DNA alignment využíván při srovnávání regulačních oblastí genů, definování genů a celogenomových srovnáváních, blízce příbuzné organizmy

12 TYPY ALIGNMENTŮ globální alignment - pokouší se nalézt nejlepší možný alignment celých sekvencí lokální alignment - pokouší se nalézt podobné úseky v sekvencích, nemusí nezbytně nalézt nejlepší alignment celých sekvencí ZVELEBIL (2006)
13 SHRNUTÍ alignment je základní a zakládající bioinformatickou metodou alignment slouží k anotaci sekvencí (funkce, evoluce, polymorfismus, motivy) alignment užitečný především když naznačuje ortologii sekvencí pod 30 % sekvenční identity je schopnost odvozovat funkci sporná, ani vysoká sekvenční identita však 100% nezaručuje úspěch.
14 JAK SE DĚLÁ ALIGNMENT?
15 KOLIK ALIGNMENTŮ LZE VYTVOŘIT? GGCATGAG GTACGTACG GTACGTACG GGCATGAGG GTACGTACG GGCATGAGG -GTACGTACG- GGC-ATG-AGG GT-ACGTACG GGCATG-AGG KTERÝ ALIGNMENT SE VÁM ZDÁ POCITOVĚ NEJSMYSLUPLNĚJŠÍ? PROČ? GTACG--T-ACG G---GCATGAGG
16 podobně u proteinů - např. Ala budu zaměněn spíš Val, Leu nebo Ile než Tyr většina by se asi shodla na předposledním alignmentu největší sekvenční identita - 0%, 33%, 22%, 50%, 42% krátký alignment, málo mezer (delece nebo inzerce obecné méně pravděpodobná než substituce) obecně G/A nebo C/T záměna pravděpodobnější než G/C nebo A/T (pyrimidin za purin)
17 ALIGNMENT - POŽADAVKY CO JE OBECNĚ POTŘEBA K NALEZENÍ NEJLEPŠÍHO ALIGNMENTU? měřítko kvality alignmentu - skóre pro shodu/ neshodu (všechny kombinace) a skóre pro mezeru (obvykle negativní - gap penalty) algoritmus, který systematicky projde všechny možné alignmenty
18 SKÓRE ALIGMENTU vychází se ze skórovací tabulky, která přiděluje určité skóre každe možné kombinaci skórovací tabulky jsou symetrické SHODA = 1 NESHODA =0 SHODA =3 NESHODA (PU/PU ; PY/PY) =0 NESHODA (PU/PY) =-3 A C G T A C G T A C G T A C G T
19 GAP PENALTY alignment score lze dobře statisticky odvodit z alignmentu, gap penalty se odhadují hůř...většinou se odhadují metodou pokusu a omylu buď konstantní - každá mezera za stejnou pokutu nebo rozdílná pokuta pro první (gap-opening penalty) a každou další mezeru (gap-extension penalty) -> affine gap penalties první mezera má výšší penaltu než každá další
20 GAP PENALTY PHE ASP ILE CYS ARG LEU PRO GLY SER ALA GLU ALA VAL CYS PHE ASN VAL CYS ARG THR PRO GLU ALA ILE CYS PHE ASN VAL CYS ARG THR PRO GLU ALA ILE CYS jedním z pokusů je definování variabilních gap penalties založených na strukturách -> nižší pravděpodobnost výskytu mezery v pravidelných sekundárních strukturách, v hydrofóbních jádrech
21 SKÓRE PRO NÁŠ ALIGNMENT GGCATGAG GTACGTACG SHODA =3 NESHODA (PU/PU ; PY/PY) =0 NESHODA (PU/PY) =-3 GAP PENALTY (KONSTANTNÍ) = -5 A C G T A C G T
22 Alignment Shoda (+3) Pu/Pu (0) Pu/Py (-3) Gap (-5) Score GTACGTACG GGCATGAGG GTACGTACG GGCATGAGG -GTACGTACG- GGC-ATG-AGG GT-ACGTACG GGCATG-AGG GTACG--T-ACG G---GCATGAGG
23 Alignment Shoda (+3) Pu/Pu (0) Pu/Py (-3) Gap (-5) Score GTACGTACG GGCATGAGG GTACGTACG GGCATGAGG -GTACGTACG- GGC-ATG-AGG GT-ACGTACG GGCATG-AGG GTACG--T-ACG G---GCATGAGG
24 A TEĎ VY! Alignment Shoda (+3) Pu/Pu (0) Pu/Py (0) Gap (-4) Score GTACGTACG GGCATGAGG GT-ACGTACG GGCATG-AGG
25 A TEĎ VY Alignment Shoda (+3) Pu/Pu (0) Pu/Py (0) Gap (-4) Score GTACGTACG GGCATGAGG GT-ACGTACG GGCATG-AGG
26 NEJLEPŠÍ ALIGNMENT - SKUTEČNÝ ALIGNMENT? alignment s nejvyšším skóre se nazývá optimální alignment, ostatní alignmenty jsou suboptimální optimální alignment však nemusí být ten který popisuje, co se v evoluci skutečně stalo naopak, ani spávný alignment nemusí mít optimální skóre nicméně, současné protokoly poskytují většinou užitečné výsledky...se zrnkem soli je však třeba je brát vždy
27 SKÓROVACÍ TABULKY (SUBSTITUTION MATRICES) používané jako základ pro výpočet skóre alignmentu DNA skórovací tabulky jednoduché - např.: shoda = +1, neshoda = 0 nebo shoda = +5, neshoda = -4 DNA sekvence se používají méně, neboť se pro většinu aplikací hodí více proteinové sekvence
28 PROTEINOVÉ SUBSTITUTION MATRICES výsledný alignment je ovlivněn zvolenou substituční tabulky, proto je jim věnována velké pozornost na začátku tabulky, které vycházely z fyzikálněchemických vlastností aminokyselin nebo z genetického kódu postupně nahrazeny tabulkami, které odpovídají skutečně pozorovaným mírám záměn mezi jednotlivými aminokyselinami

29 PAM PAM = Percent Accepted Mutations vyvinuta už v 70. letech 20. století Margaretou Dayhoff založena na pravděpodobných mírách mutace kalkulovaných z globálních alignmentů blízce podobných sekvencí kalkulována jako logaritmus pravděpodobnosti záměny aminokyseliny log odd = počet pozorovaných záměn/ počet očekávaných záměň při náhodném zaměňování
30 KALKULACE PRAVDĚPODOBNOSTI skóre pro záměnu valinu za isoleucin pravděpodobnost záměny pokud jsou sekvence příbuzné (pozorovaná) = 0.03 výskyt aminokyselin v populaci (databáze, proteom) = 0.1 a 0.05 poměr pravděpodobností = 0.03/(0.1*0.05) = 6x větší pravděpodobnost záměny V -> I než očekáváno při náhodných záměnách skóre = desítkový logaritmus * 10 a zaokrouhlen na nejbližší celé číslo = 10*log 6 = 10 * = 8
 založeno na lokálních alignmentech bloků")

31 BLOSUM BLOSUM = BLOck SUbstitution Matrix vytvořeny na začátku 90. let (Henikoff a Henikoff, 1992) založeno na lokálních alignmentech bloků aminokyselin s definovanou sekvenční identitou (u homologních proteinů) všechny tabulky vycházejí z experimentální dat, nejsou extrapolovány jako některé PAM tabulky


32 BLOSUM BLOSUM 80 - tabulka vypočítaná na základě alignmentu bloku sekvencí s identitou 80 % BLOSUM 62 - tabulka vypočítaná na základě alignmentu bloku sekvencí s identitou 62 %
33 BLOSUM VERSUS PAM PAM BLOSUM z globálního alignmentu velmi podobných sekvencí hodnoty pro vzdáleně podobné sekvence extrapolovány z hodnot pro velmi podobné sekvence evoluční vzdálenost z lokálního alignmentu bez mezer všechny hodnoty vycházejí ze skutečných alignmentů sekvenční identita
34 DNASEKVENCE RNASEKVENCE PAM = 44 BLOSUM = 51
35 ALIGNMENT - POŽADAVKY CO JE OBECNĚ POTŘEBA K NALEZENÍ NEJLEPŠÍHO ALIGNMENTU? měřítko kvality alignmentu - skóre pro shodu/ neshodu (všechny kombinace) a skóre pro mezeru (obvykle negativní - gap penalty) algoritmus, který systematicky projde všechny možné alignmenty
36 počet alignmentů roste pokud povolíme mezery generovat všechny možné alignmenty není praktické, neboť dvě sekvence o 100 aminokyselinách mají asi možných alignmentů současné stolní počítače zvládnou okolo 2,5 x 10 9 operací za vteřinu (1 rok = vteřin) x 2,5 x 10 9 ~ = 8 x operací/rok máme i rychlejší počítače, ale...
37 NEEDLEMAN-WUNSCH řešení přinesl až algoritmus Needlemana a Wunsche (1970), který modifikoval v matematice a programování používané algoritmy dynamického programování (dynamic programming) dynamic programming vyvinuto ve 40. letech 20. století jako způsob řešení problémů, kde se dělá řada rozhodnutí krok po kroku programming nemá spojení s programováním ve smyslu psaní programů, ale v matematickém smyslu jako optimizace průběh algoritmu se mění s konkrétním problémem, a proto je dynamický
38 pro každou pozici v alignmentu počítá s pomocí scoring matrix, zda je v daném místě optimálnější (vyšší skóre), aby došlo k shodě/neshodě v sekvenci či inzerci nebo deleci Needleman-Wunsch algoritmus dovede v každém kroku zavrhnout řadu alignmentů zvládnutelné v reálném čase
39 JAK TO FUNGUJE? jednu sekvenci píšeme do řádku, druhou do sloupce tabulky pro každou pozici v tabulce se počítá pravděpodobnost třech událostí: shody (záměny), inzerce v první sekvenci a inzerce v druhé sekvenci DI-1,J-1 DI,J-Y DI-1,J-1 + B (AJ, BJ) DI,J-Y - W (Y) DI-X,J DI-X,J - W (X)
40 JAK TO FUNGUJE? GAATTCAGTTA GGATCGA SKÓROVACÍ TABULKA: SI,J = 1 (POKUD SHODA) SI,J = 0 (POKUD NESHODA) W = 0 (GAP PENALTY) A C G T A C G T
41 ZAČÍNÁME VYPLŇOVAT G A A T T C A G T T A G 0 G 0 A 0 T 0 C 0 G 0 A 0
42 G A A T T C A G T T A G 0 1 G 0 A 0 T 0 C 0 G 0 A 0 DI,J-Y DI,J-Y - W (Y) DI-1,J-1 + B (AJ, BJ) DI-X,J - W (X)
43 G A A T T C A G T T A G G 0 1 A 0 1 T 0 1 C 0 1 G 0 1 A 0 1
44 G A A T T C A G T T A G G A T 0 1 C 0 1 G 0 1 A 0 1
45 DOPLNÍME TABULKU G A A T T C A G T T A G G A T C G A
46 HLEDÁME ALIGNMENT G A A T T C A G T T A G G A T C G A
47 HLEDÁME ALIGNMENT G A A T T C A G T T A G G A T C G A SEKVENCE 1 A SEKVENCE 2 A
48 HLEDÁME ALIGNMENT G A A T T C A G T T A G G A T C G A SEKVENCE 1 TA SEKVENCE 2 -A
49 ŘEŠENÍ G A A T T C A G T T A G G A T C G A SEKVENCE 1 GAATTCAGTTA SEKVENCE 2 GGA-TC-G--A
50 HLEDÁNÍ ALIGNMENTU -SHRNUTÍ stopujeme alignment zpětně pro každou pozici určujeme odkuď jsme se k ní dostali v některých případech může vzniknout více optimálních alignmentů, program však zobrazí pouze jedno řešení
51 NEEDLEMAN-WUNSCH SHRNUTÍ využívá dynamické programování k zredukování možných alignmentů vytváří globální alignment zaručuje nalezení optimálního alignmentu (s přihlédnutím ke zvolené skórovací tabulce a gap penalty )

52 MULTIPLE SEQUENCE ALIGNMENT (MSA)
53 PROČ MSA? pokud srovnáváme příbuzné proteiny tak dostáváme silnější signál umožnuje nám zpřesnit alignment vzniklý srovnáním dvou sekvencí větší šance nalezení strukturně nebo funkčně významných aminokyselin základ pro fylogenetické studie

54 JAK NA MSA? lze použít dynamické programování, ale počet rozměrů v matrici roste úměrně s počtem srovnávaných sekvencí pokud by alignment 2 sekvencí o 50 aminokyselinách trval vteřinu, pak srovnání N sekvencí by trvalo 10 2N-4 s - 3 sekvence ~2 minuty, 4 sekvence ~ 3 hodiny, 5 sekvencí ~ 11,5 dne dynamické programování je pro více jak sekvence příliš pomalé tři a proto jej žádný z běžných programů nepoužívá
55 JAK NA MSA? většina programů používá hierarchické progresivní metody všechny kombinace sekvencí projdou pairwise sequence alignment alignmenty jsou hierarchicky seřazeny dle míry podobnosti (fylogenetický strom) finální multiple alignment je budován v krocích -první jsou seřazeny nejpodobnější sekvence, k takovému páru se přirovnává další nejbližší sekvence dokud nejsou použity všechny sekvence
 finální multiple alignment je budován v krocích")
56 hierarchické progresivní metody MSA PŘÍSTUPY? všechny kombinace sekvencí projdou pairwise sequence alignment alignmenty jsou hierarchicky seřazeny dle míry podobnosti (fylogenetický strom) finální multiple alignment je budován v krocích -první jsou seřazeny nejpodobnější sekvence, k takovému páru se přirovnává další nejbližší sekvence dokud nejsou použity všechny sekvence Clustal W, T-cofee

57 CLUSTAL W VŠECHNY ALIGNMENTY MÍRA PODOBNOSTI SEKVENCÍ GUIDE TREE POSTUPNÉ SROVNÁVÁNÍ THOMPSON ET AL., 1994
58 MSA V PRAXI největší slabinou je, že chyby vytvořené v úvodních alignmentech se propagují do výsledného alignmentu iterativní metody - optimizace objektivní funkce přes realigning podskupin sekvencí - Muscle, ProbCons učící metody - HMM, genetické algoritmy, simulated annealing - FSA phylogeny-aware methods - PRANK, PAGAN MSA porovnávány proti databázi strukturních alignmentů (BALiBase)


59
60 MAFFT metoda pro velké soubory dat (fylogenetické analýzy) homologické oblasti jsou identifikovány pomocí rychlých Fourierovách transformací (objem a polarita aa) alignment kombinací progresivních a iterativních metod až 100x rychlejší než T-cofee při stejné přesnosti novější FAMSA (Deorowicz, 2016)

61 PHYLOGENY-AWARE MSA PAGAN, ProGraphMSA snaží se minimalizovat počet mezer v alignmentu
62 FAST STATISTICAL ALIGNMENT používá machine learning metodu simulated annealing na základě pairwise alignmentů gap penalties i skorovací tabulky jsou odhadovány pro každý set sekvencí individuálně - velmi robustní i pro velmi dlouhé sekvence
63 STRUKTURNÍ / SEKVENČNÍ ALIGNMENT struktura lépe konzervována než sekvence umožňuje odhalovat homologie v sekvenční twilight (midnight) zone vylepšuje multiple sequence alignments, predikce struktury a metodiku alignmentu obecně
64 STRUKTURNÍ VERSUS SEKVENČNÍ 1FWR_A 2YPI_A 1FWR_A 2YPI_A 1FWR_A 2YPI_A 1FWR_A 2YPI_A MKNWKTSAESILTTGPVVPVIVVKKLEHAVPMAKA ARTFFVGGNFKLNGSKQSIKEIVERLNTASIPENVEVVICPPATYLDYSVSLVKKPQVTV ::... : :. *.. :. *... LVAGGVRVLEVTLRTECAVDAIRAIAKEVPEAIVGAGTVLNPQQLAEVTE AGA GAQNAYLKASGAFTGENSVDQIKDVGAKWVILGHSERRSYFHEDDKFIADKTKFALGQGV.... :: * :** *: :. :. :: ::: *. QFAISPGLTEPLLKAATEGTIPLIPGISTVSELMLGMDYGLKEFQFFPAEANGGVKALQA GVILCIGETLEEKKAGKTLDVVERQLNAVLEEVKDWTNVVVAYEPVWAIGTGLAATPEDA. :. * * **.. : :.:.*: : :.:. :.... :* IAGPFSQVRFCPKGGISPANYRDYLALKSVLCIGGSWLVPADALEAGDYDRITKLAREAV QDIHASIRKFLASKLGDKAASELRILYGGSANGSNAVTFKDKADVDGFLVGGASLKPEFV * :*... *. :...:..* * :.* * *
65 STRUKTURNÍ VERSUS SEKVENČNÍ Sequence ART---FFVGGNFKLNG-SKQSI-KEIVERLNTASI--PENVEVVICP.=ALI =ID Sequence 2 MKNWKTSAESIL--TTGP--VVPVI--VVKKLEHAVP-MAKALVAG-GVR-----V-LEV Sequence PATYLDYSVSLV-KKPQVTVGAQ-N--AY-LKASGAFTGEN-S---VDQIKDVG.=ALI =ID Sequence 2 TLRTECAVDAIRAIAKEVP-E--AIVGAGTVLN-PQ QLAEVT--E---AG Sequence 1 AKWVILGH--SERRSYFHEDDKFIADKTKFALGQGVGVILCIGETLEEKKAGKTLDVVER.=ALI =ID Sequence 2 AQFAIS-PGL TEPLLKAATEGTIPLIPGIS TVS Sequence 1 QLNAV-LEEVKDW-TNVVVAYEP--VW--AIGTGLAATPEDA--QDI--HASI-RKFLA-.=ALI =ID Sequence 2 ELMLGMD--YG-LK---EFQFFPAE-ANG G----VKA--LQA--IAG-P--FS Sequence 1 SKLGDKAA-SELRILYGGSANGSN-AVTF---KDK-ADVDGFLVGGA-SLK =ALI =ID Sequence QV---RFCPKGGIS-PANY--RDYL--ALKSVLCIGG-SWL-VPADALEAGDY

66 STRUKTURNÍ VERSUS SEKVENČNÍ
67 KVALITA ALIGNMETU lze hodnotit za pomoci strukturní informace MSA porovnávány proti databázi strukturních alignmentů (BALiBase), HomFam program APDB (součást T-Coffee) pokud jsou známy alespoň dvě struktury umožňuje vybrat nejlepší z alternativních alignmentů QuanTest (2017) - za pomoci přesnosti predikce sekundárních struktur
 pro účely fylogenetických analýz se často vyřazují oblasti se spoustou mezer TrimAl,")
68 není uniformní KVALITA UVNITŘ ALIGNMENTU MSA programy však často neoznačují kterým částem alignmentu věří a kterým nikoliv (FSA) pro účely fylogenetických analýz se často vyřazují oblasti se spoustou mezer TrimAl, JalView, UGENE
69 STROM ZE VŠECH GENOMOVÝCH SEKVENCÍ výběr genů, identifikace ortologů.. whole-genome phylogenies většinou alignment-free metody - pořadí genů, množství genů, nukleotidové složení, SNP, metabolické dráhy -> kompletně pomíjí evoluční koncepty alignment metody spoléhající na průměrné hodnoty podobností - typicky pracují jen z ortology plně automatické metody používající i neortologické sekvence (Yonoko et al., 2018)
70 SHRNUTÍ databáze by měly být pravidelně updatovány přehled dostupných biologických databází vždy v lednovém čísle NAR řada velmi specializovaných databází hledání v databázích povětšinou heuristickými metodami standard dnes BLAST nutno hodnotit statistickou významnost nálezu citlivější metodou PSI-Blast nebo HMM metody
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
Thursday, February 27, 14
 DATABÁZE A VYHLEDÁVÁNÍ SEKVENCÍ MOLEKULÁRNÍ TAXONOMIE 2014 MARIAN NOVOTNÝ PŘEDNÁŠEJÍCÍ Mgr. Marian NOVOTNÝ, PhD. vystudoval odbornou biologii na PřF UK, diplomka v laboratoři doc. Folka doktorát na Uppsalské
DATABÁZE A VYHLEDÁVÁNÍ SEKVENCÍ MOLEKULÁRNÍ TAXONOMIE 2014 MARIAN NOVOTNÝ PŘEDNÁŠEJÍCÍ Mgr. Marian NOVOTNÝ, PhD. vystudoval odbornou biologii na PřF UK, diplomka v laboratoři doc. Folka doktorát na Uppsalské
Základy genomiky. I. Úvod do bioinformatiky. Jan Hejátko
 Základy genomiky I. Úvod do bioinformatiky Jan Hejátko Masarykova univerzita, Oddělení funkční genomiky a proteomiky Laboratoř molekulární fyziologie rostlin Základy genomiky I. Zdrojová literatura ke
Základy genomiky I. Úvod do bioinformatiky Jan Hejátko Masarykova univerzita, Oddělení funkční genomiky a proteomiky Laboratoř molekulární fyziologie rostlin Základy genomiky I. Zdrojová literatura ke
Studijní materiály pro bioinformatickou část ViBuChu. úloha II. Jan Komárek, Gabriel Demo
 Studijní materiály pro bioinformatickou část ViBuChu úloha II Jan Komárek, Gabriel Demo Adenin Struktura DNA Thymin 5 konec 3 konec DNA tvořena dvěmi řetězci orientovanými antiparalelně (liší se orientací
Studijní materiály pro bioinformatickou část ViBuChu úloha II Jan Komárek, Gabriel Demo Adenin Struktura DNA Thymin 5 konec 3 konec DNA tvořena dvěmi řetězci orientovanými antiparalelně (liší se orientací
Využití metod strojového učení v bioinformatice David Hoksza
 Využití metod strojového učení v bioinformatice David Hoksza SIRET Research Group Katedra softwarového inženýrství, Matematicko-fyzikální fakulta Karlova Univerzita v Praze Bioinformatika Biologické inspirace
Využití metod strojového učení v bioinformatice David Hoksza SIRET Research Group Katedra softwarového inženýrství, Matematicko-fyzikální fakulta Karlova Univerzita v Praze Bioinformatika Biologické inspirace
Genomické databáze. Shlukování proteinových sekvencí. Ivana Rudolfová. školitel: doc. Ing. Jaroslav Zendulka, CSc.
 Genomické databáze Shlukování proteinových sekvencí Ivana Rudolfová školitel: doc. Ing. Jaroslav Zendulka, CSc. Obsah Proteiny Zdroje dat Predikce struktury proteinů Cíle disertační práce Vstupní data
Genomické databáze Shlukování proteinových sekvencí Ivana Rudolfová školitel: doc. Ing. Jaroslav Zendulka, CSc. Obsah Proteiny Zdroje dat Predikce struktury proteinů Cíle disertační práce Vstupní data
Inovace studia molekulární a buněčné biologie
 Inovace studia molekulární a buněčné biologie Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky. MBIO1/Molekulární biologie 1 Tento projekt je spolufinancován
Inovace studia molekulární a buněčné biologie Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky. MBIO1/Molekulární biologie 1 Tento projekt je spolufinancován
Hemoglobin a jemu podobní... Studijní materiál. Jan Komárek
 Hemoglobin a jemu podobní... Studijní materiál Jan Komárek Bioinformatika Bioinformatika je vědní disciplína, která se zabývá metodami pro shromážďování, analýzu a vizualizaci rozsáhlých souborů biologických
Hemoglobin a jemu podobní... Studijní materiál Jan Komárek Bioinformatika Bioinformatika je vědní disciplína, která se zabývá metodami pro shromážďování, analýzu a vizualizaci rozsáhlých souborů biologických
Dynamic programming. Historie. Dynamické programování je obsaženo v těchto programech: Příklad: chceme optimálně přiložit dvě sekvence
 Dynamic programming Dynamické programování je obsaženo v těchto programech: BLS FS lustalw HMMER enscan MFold Phylip Historie 9s matematik Richard Bellman při optimalizaci rozhodovacích procesů chtěl zmást
Dynamic programming Dynamické programování je obsaženo v těchto programech: BLS FS lustalw HMMER enscan MFold Phylip Historie 9s matematik Richard Bellman při optimalizaci rozhodovacích procesů chtěl zmást
Bioinformatika a výpočetní biologie KFC/BIN. I. Přehled
 Bioinformatika a výpočetní biologie KFC/BIN I. Přehled RNDr. Karel Berka, Ph.D. Univerzita Palackého v Olomouci Definice bioinformatiky (Molecular) bio informatics: bioinformatics is conceptualising biology
Bioinformatika a výpočetní biologie KFC/BIN I. Přehled RNDr. Karel Berka, Ph.D. Univerzita Palackého v Olomouci Definice bioinformatiky (Molecular) bio informatics: bioinformatics is conceptualising biology
Vyhledávání podobných sekvencí BLAST
 Vyhledávání podobných sekvencí BLAST Základní informace Následující text je součástí učebních textů předmětu Analýza sekvencí DNA a je určen hlavně pro studenty Matematické biologie. Může být ovšem přínosný
Vyhledávání podobných sekvencí BLAST Základní informace Následující text je součástí učebních textů předmětu Analýza sekvencí DNA a je určen hlavně pro studenty Matematické biologie. Může být ovšem přínosný
Evropský sociální fond Praha & EU: Investujeme do vaší budoucnosti URČOVÁNÍ PRIMÁRNÍ STRUKTURY BÍLKOVIN
 Evropský sociální fond Praha & EU: Investujeme do vaší budoucnosti URČOVÁNÍ PRIMÁRNÍ STRUKTURY BÍLKOVIN Primární struktura primární struktura bílkoviny je dána pořadím AK jejích polypeptidových řetězců
Evropský sociální fond Praha & EU: Investujeme do vaší budoucnosti URČOVÁNÍ PRIMÁRNÍ STRUKTURY BÍLKOVIN Primární struktura primární struktura bílkoviny je dána pořadím AK jejích polypeptidových řetězců
6. Kde v DNA nalézáme rozdíly, zodpovědné za obrovskou diverzitu života?
 6. Kde v DNA nalézáme rozdíly, zodpovědné za obrovskou diverzitu života? Pamatujete na to, co se objevilo v pracích Charlese Darwina a Alfreda Wallace ohledně vývoje druhů? Aby mohl mechanismus přírodního
6. Kde v DNA nalézáme rozdíly, zodpovědné za obrovskou diverzitu života? Pamatujete na to, co se objevilo v pracích Charlese Darwina a Alfreda Wallace ohledně vývoje druhů? Aby mohl mechanismus přírodního
Molekulární genetika IV zimní semestr 6. výukový týden ( )
") Ústav biologie a lékařské genetiky 1.LF UK a VFN, Praha Molekulární genetika IV zimní semestr 6. výukový týden (5.11. 9.11.2007) Nondisjunkce u Downova syndromu 2 Tři rodokmeny rodin s dětmi postiženými
Ústav biologie a lékařské genetiky 1.LF UK a VFN, Praha Molekulární genetika IV zimní semestr 6. výukový týden (5.11. 9.11.2007) Nondisjunkce u Downova syndromu 2 Tři rodokmeny rodin s dětmi postiženými
DATABÁZE A VYHLEDÁVÁNÍ SEKVENCÍ
 DATABÁZE A VYHLEDÁVÁNÍ SEKVENCÍ MOLEKULÁRNÍ TAXONOMIE 2015 MARIAN NOVOTNÝ OSNOVA co je substrát pro molekulární taxonomii? kde se shromažďují data? jak data vyhledávat? REKONSTRUKCE EVOLUČNÍ HISTORIE rekonstrukce
DATABÁZE A VYHLEDÁVÁNÍ SEKVENCÍ MOLEKULÁRNÍ TAXONOMIE 2015 MARIAN NOVOTNÝ OSNOVA co je substrát pro molekulární taxonomii? kde se shromažďují data? jak data vyhledávat? REKONSTRUKCE EVOLUČNÍ HISTORIE rekonstrukce
Využití strojového učení k identifikaci protein-ligand aktivních míst
 Využití strojového učení k identifikaci protein-ligand aktivních míst David Hoksza, Radoslav Krivák SIRET Research Group Katedra softwarového inženýrství, Matematicko-fyzikální fakulta Karlova Univerzita
Využití strojového učení k identifikaci protein-ligand aktivních míst David Hoksza, Radoslav Krivák SIRET Research Group Katedra softwarového inženýrství, Matematicko-fyzikální fakulta Karlova Univerzita
Vytváření fylogenetických stromů na základě alignmentů. Tomáš Novotný Jaroslav Knotek
 Vytváření fylogenetických stromů na základě alignmentů Tomáš Novotný Jaroslav Knotek Alignmenty - opakování Existují dvě základní varianty alignmentů: globální a lokální Globální: Hledáme nejlepší zarovnání
Vytváření fylogenetických stromů na základě alignmentů Tomáš Novotný Jaroslav Knotek Alignmenty - opakování Existují dvě základní varianty alignmentů: globální a lokální Globální: Hledáme nejlepší zarovnání
b) Jak se změní sekvence aminokyselin v polypeptidu, pokud dojde v pozici 23 k záměně bázového páru GC za TA (bodová mutace) a s jakými následky?
 Jak se změní sekvence aminokyselin v polypeptidu, pokud dojde v pozici 23 k záměně bázového páru GC za TA (bodová mutace) a s jakými následky?") 1.1: Gén pro polypeptid, který je součástí peroxidázy buku lesního, má sekvenci 3'...TTTACAGTCCATTCGACTTAGGGGCTAAGGTACCTGGAGCCCACGTTTGGGTCATCCAG...5' 5'...AAATGTCAGGTAAGCTGAATCCCCGATTCCATGGACCTCGGGTGCAAACCCAGTAGGTC...3'
1.1: Gén pro polypeptid, který je součástí peroxidázy buku lesního, má sekvenci 3'...TTTACAGTCCATTCGACTTAGGGGCTAAGGTACCTGGAGCCCACGTTTGGGTCATCCAG...5' 5'...AAATGTCAGGTAAGCTGAATCCCCGATTCCATGGACCTCGGGTGCAAACCCAGTAGGTC...3'
Molekulárn. rní genetika
 Molekulárn rní genetika Centráln lní dogma molekulárn rní biologie cesta přenosu genetické informace: DNA RNA proteiny výjimkou reverzní transkripce retrovirů: RNA DNA Chemie nukleových kyselin dusíkaté
Molekulárn rní genetika Centráln lní dogma molekulárn rní biologie cesta přenosu genetické informace: DNA RNA proteiny výjimkou reverzní transkripce retrovirů: RNA DNA Chemie nukleových kyselin dusíkaté
Aminokyseliny. Gymnázium a Jazyková škola s právem státní jazykové zkoušky Zlín. Tematická oblast Datum vytvoření Ročník Stručný obsah Způsob využití
 Aminokyseliny Tematická oblast Datum vytvoření Ročník Stručný obsah Způsob využití Autor Kód Chemie přírodních látek proteiny 18.7.2012 3. ročník čtyřletého G Určování postranních řetězců aminokyselin
Aminokyseliny Tematická oblast Datum vytvoření Ročník Stručný obsah Způsob využití Autor Kód Chemie přírodních látek proteiny 18.7.2012 3. ročník čtyřletého G Určování postranních řetězců aminokyselin
Využití DNA markerů ve studiu fylogeneze rostlin
 Mendelova genetika v příkladech Využití DNA markerů ve studiu fylogeneze rostlin Ing. Petra VESELÁ Ústav lesnické botaniky, dendrologie a geobiocenologie LDF MENDELU Brno Tento projekt je spolufinancován
Mendelova genetika v příkladech Využití DNA markerů ve studiu fylogeneze rostlin Ing. Petra VESELÁ Ústav lesnické botaniky, dendrologie a geobiocenologie LDF MENDELU Brno Tento projekt je spolufinancován
DATABÁZE A VYHLEDÁVÁNÍ SEKVENCÍ
 DATABÁZE A VYHLEDÁVÁNÍ SEKVENCÍ MOLEKULÁRNÍ TAXONOMIE 2015 MARIAN NOVOTNÝ OSNOVA co je substrát pro molekulární taxonomii? kde se shromažďují data? jak data vyhledávat? REKONSTRUKCE EVOLUČNÍ HISTORIE rekonstrukce
DATABÁZE A VYHLEDÁVÁNÍ SEKVENCÍ MOLEKULÁRNÍ TAXONOMIE 2015 MARIAN NOVOTNÝ OSNOVA co je substrát pro molekulární taxonomii? kde se shromažďují data? jak data vyhledávat? REKONSTRUKCE EVOLUČNÍ HISTORIE rekonstrukce
Proteiny Genová exprese. 2013 Doc. MVDr. Eva Bártová, Ph.D.
 Proteiny Genová exprese 2013 Doc. MVDr. Eva Bártová, Ph.D. Bílkoviny (proteiny), 15% 1g = 17 kj Monomer = aminokyseliny aminová skupina karboxylová skupina α -uhlík postranní řetězec Znát obecný vzorec
Proteiny Genová exprese 2013 Doc. MVDr. Eva Bártová, Ph.D. Bílkoviny (proteiny), 15% 1g = 17 kj Monomer = aminokyseliny aminová skupina karboxylová skupina α -uhlík postranní řetězec Znát obecný vzorec
Počítačové vyhledávání genů a funkčních oblastí na DNA
 Počítačové vyhledávání genů a funkčních oblastí na DNA Hodnota genomových sekvencí záleží na kvalitě anotace Anotace Charakterizace genomových vlastností s použitím výpočetních a experimentálních metod
Počítačové vyhledávání genů a funkčních oblastí na DNA Hodnota genomových sekvencí záleží na kvalitě anotace Anotace Charakterizace genomových vlastností s použitím výpočetních a experimentálních metod
Faculty of Nuclear Sciences and Physical Engineering Czech Technical University in Prague
 1 / 23 Faculty of Nuclear Sciences and Physical Engineering Czech Technical University in Prague 2 / 23 biologové často potřebují najít často se opakující sekvence DNA tyto sekvence bývají relativně krátké,
1 / 23 Faculty of Nuclear Sciences and Physical Engineering Czech Technical University in Prague 2 / 23 biologové často potřebují najít často se opakující sekvence DNA tyto sekvence bývají relativně krátké,
Molekulární biotechnologie č.9. Cílená mutageneze a proteinové inženýrství
 Molekulární biotechnologie č.9 Cílená mutageneze a proteinové inženýrství Gen kódující jakýkoliv protein lze izolovat z přírody, klonovat, exprimovat v hostitelském organismu. rekombinantní protein purifikovat
Molekulární biotechnologie č.9 Cílená mutageneze a proteinové inženýrství Gen kódující jakýkoliv protein lze izolovat z přírody, klonovat, exprimovat v hostitelském organismu. rekombinantní protein purifikovat
Bioinformatika pro PrfUK 2003
 Bioinformatika pro PrfUK 2003 Jiří Vondrášek Ústav organické chemie a biochemie vondrasek@uochb.cas.cz Jan Pačes Ústav molekulární genetiky hpaces@img.cas.cz http://bio.img.cas.cz/prfuk2003 What is Bioinformatics?---The
Bioinformatika pro PrfUK 2003 Jiří Vondrášek Ústav organické chemie a biochemie vondrasek@uochb.cas.cz Jan Pačes Ústav molekulární genetiky hpaces@img.cas.cz http://bio.img.cas.cz/prfuk2003 What is Bioinformatics?---The
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/
 Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky Populační genetika (KBB/PG)
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky Populační genetika (KBB/PG)
Bioinformatika. Jiří Vondrášek Ústav organické chemie a biochemie Jan Pačes Ústav molekulární genetiky
 Bioinformatika pro PrfUK 2006 Jiří Vondrášek Ústav organické chemie a biochemie vondrasek@uochb.cas.cz Jan Pačes Ústav molekulární genetiky hpaces@img.cas.cz http://bio.img.cas.cz/prfuk2006 syllabus Úterý,
Bioinformatika pro PrfUK 2006 Jiří Vondrášek Ústav organické chemie a biochemie vondrasek@uochb.cas.cz Jan Pačes Ústav molekulární genetiky hpaces@img.cas.cz http://bio.img.cas.cz/prfuk2006 syllabus Úterý,
Aplikovaná bioinformatika
 Aplikovaná bioinformatika Číslo aktivity: 2.V Název klíčové aktivity: Na realizaci se podílí: Implementace nových předmětů do daného studijního programu doc. RNDr. Michaela Wimmerová, Ph.D., Mgr. Josef
Aplikovaná bioinformatika Číslo aktivity: 2.V Název klíčové aktivity: Na realizaci se podílí: Implementace nových předmětů do daného studijního programu doc. RNDr. Michaela Wimmerová, Ph.D., Mgr. Josef
METODY VÍCENÁSOBNÉHO ZAROVNÁVÁNÍ NUKLEOTIDOVÝCH SEKVENCÍ
 VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA ELEKTROTECHNIKY A KOMUNIKAČNÍCH TECHNOLOGIÍ ÚSTAV BIOMEDICÍNSKÉHO INŽENÝRSTVÍ FACULTY OF ELECTRICAL ENGINEERING AND COMMUNICATION DEPARTMENT
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA ELEKTROTECHNIKY A KOMUNIKAČNÍCH TECHNOLOGIÍ ÚSTAV BIOMEDICÍNSKÉHO INŽENÝRSTVÍ FACULTY OF ELECTRICAL ENGINEERING AND COMMUNICATION DEPARTMENT
Základy fylogenetiky a konstrukce fylogenetických stromů
 EKO/MEM Molekulární ekologie mikroorganismů Základy fylogenetiky a konstrukce fylogenetických stromů Iva Buriánková Katedra ekologie PřF UP Kde vyrostl první fylogenetický strom? Charles Darwin (1809 1882)
EKO/MEM Molekulární ekologie mikroorganismů Základy fylogenetiky a konstrukce fylogenetických stromů Iva Buriánková Katedra ekologie PřF UP Kde vyrostl první fylogenetický strom? Charles Darwin (1809 1882)
Virtuální svět genetiky 1. Translace
 (překlad) je druhým krokem exprese genetické informace a ukončuje dráhu DNA > RNA > protein. probíhá mimo jádro, v cytoplazmě na ribozómech. Výchozími látkami pro translaci je 21 standardních aminokyselin,
(překlad) je druhým krokem exprese genetické informace a ukončuje dráhu DNA > RNA > protein. probíhá mimo jádro, v cytoplazmě na ribozómech. Výchozími látkami pro translaci je 21 standardních aminokyselin,
Metody studia historie populací. Metody studia historie populací
 1) Metody studia genetické rozmanitosti komplexní fenotypové znaky, molekulární znaky. 2) Mechanizmy evoluce mutace, přírodní výběr, genový posun a genový tok 3) Anageneze x kladogeneze - co je vlastně
1) Metody studia genetické rozmanitosti komplexní fenotypové znaky, molekulární znaky. 2) Mechanizmy evoluce mutace, přírodní výběr, genový posun a genový tok 3) Anageneze x kladogeneze - co je vlastně
Pravděpodobnost v závislosti na proměnné x je zde modelován pomocí logistického modelu. exp x. x x x. log 1
 Logistická regrese Menu: QCExpert Regrese Logistická Modul Logistická regrese umožňuje analýzu dat, kdy odezva je binární, nebo frekvenční veličina vyjádřená hodnotami 0 nebo 1, případně poměry v intervalu
Logistická regrese Menu: QCExpert Regrese Logistická Modul Logistická regrese umožňuje analýzu dat, kdy odezva je binární, nebo frekvenční veličina vyjádřená hodnotami 0 nebo 1, případně poměry v intervalu
Genetický kód. Jakmile vznikne funkční mrna, informace v ní obsažená může být ihned použita pro syntézu proteinu.
 Genetický kód Jakmile vznikne funkční, informace v ní obsažená může být ihned použita pro syntézu proteinu. Pravidla, kterými se řídí prostřednictvím přenos z nukleotidové sekvence DNA do aminokyselinové
Genetický kód Jakmile vznikne funkční, informace v ní obsažená může být ihned použita pro syntézu proteinu. Pravidla, kterými se řídí prostřednictvím přenos z nukleotidové sekvence DNA do aminokyselinové
"Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Molekulární základy genetiky
 "Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Molekulární základy genetiky 1/76 GENY Označení GEN se používá ve dvou základních významech: 1. Jako synonymum pro vlohu
"Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Molekulární základy genetiky 1/76 GENY Označení GEN se používá ve dvou základních významech: 1. Jako synonymum pro vlohu
Genetický polymorfismus
 Genetický polymorfismus Za geneticky polymorfní je považován znak s nejméně dvěma geneticky podmíněnými variantami v jedné populaci, které se nachází v takových frekvencích, že i zřídkavá má frekvenci
Genetický polymorfismus Za geneticky polymorfní je považován znak s nejméně dvěma geneticky podmíněnými variantami v jedné populaci, které se nachází v takových frekvencích, že i zřídkavá má frekvenci
Cysteinové adukty globinu jako potenciální biomarkery expozice styrenu
 Cysteinové adukty globinu jako potenciální biomarkery expozice styrenu J. Mráz, I. Hanzlíková, Š. Dušková, E. Frantík, V. Stránský Státní zdravotní ústav Praha 1 Biomarkery expozice cizorodým látkám výchozí
Cysteinové adukty globinu jako potenciální biomarkery expozice styrenu J. Mráz, I. Hanzlíková, Š. Dušková, E. Frantík, V. Stránský Státní zdravotní ústav Praha 1 Biomarkery expozice cizorodým látkám výchozí
Využití DNA sekvencování v
 Využití DNA sekvencování v taxonomii prokaryot Mgr. Pavla Holochová, doc. RNDr. Ivo Sedláček, CSc. Česká sbírka mikroorganismů Ústav experimentální biologie Přírodovědecká fakulta Masarykova univerzita,
Využití DNA sekvencování v taxonomii prokaryot Mgr. Pavla Holochová, doc. RNDr. Ivo Sedláček, CSc. Česká sbírka mikroorganismů Ústav experimentální biologie Přírodovědecká fakulta Masarykova univerzita,
Populační genetika. ) a. Populační genetika. Castle-Hardy-Weinbergova zákonitost. Platí v panmiktické populaci za předpokladu omezujících podmínek
 a. Populační genetika. Castle-Hardy-Weinbergova zákonitost. Platí v panmiktické populaci za předpokladu omezujících podmínek") Poulační genetika Poulační genetika ORGANISMUS Součást výše organizované soustavy oulace POPULACE Soubor jedinců jednoho druhu Genotyově heterogenní V určitém čase má řirozeně vymezený rostor Velký očet
Poulační genetika Poulační genetika ORGANISMUS Součást výše organizované soustavy oulace POPULACE Soubor jedinců jednoho druhu Genotyově heterogenní V určitém čase má řirozeně vymezený rostor Velký očet
Markovovy modely v Bioinformatice
 Markovovy modely v Bioinformatice Outline Markovovy modely obecně Profilové HMM Další použití HMM v Bioinformatice Analýza biologických sekvencí Biologické sekvence: DNA,RNA,protein prim.str. Sekvenování
Markovovy modely v Bioinformatice Outline Markovovy modely obecně Profilové HMM Další použití HMM v Bioinformatice Analýza biologických sekvencí Biologické sekvence: DNA,RNA,protein prim.str. Sekvenování
Struktura proteinů. - testík na procvičení. Vladimíra Kvasnicová
 Struktura proteinů - testík na procvičení Vladimíra Kvasnicová Mezi proteinogenní aminokyseliny patří a) kyselina asparagová b) kyselina glutarová c) kyselina acetoctová d) kyselina glutamová Mezi proteinogenní
Struktura proteinů - testík na procvičení Vladimíra Kvasnicová Mezi proteinogenní aminokyseliny patří a) kyselina asparagová b) kyselina glutarová c) kyselina acetoctová d) kyselina glutamová Mezi proteinogenní
Profilování vzorků heroinu s využitím vícerozměrné statistické analýzy
 Profilování vzorků heroinu s využitím vícerozměrné statistické analýzy Autor práce : RNDr. Ivo Beroun,CSc. Vedoucí práce: prof. RNDr. Milan Meloun, DrSc. PROFILOVÁNÍ Profilování = klasifikace a rozlišování
Profilování vzorků heroinu s využitím vícerozměrné statistické analýzy Autor práce : RNDr. Ivo Beroun,CSc. Vedoucí práce: prof. RNDr. Milan Meloun, DrSc. PROFILOVÁNÍ Profilování = klasifikace a rozlišování
Mutace jako změna genetické informace a zdroj genetické variability
 Obecná genetika Mutace jako změna genetické informace a zdroj genetické variability Doc. RNDr. Ing. Eva PALÁTOVÁ, PhD. Ing. Roman LONGAUER, CSc. Ústav zakládání a pěstění lesů LDF MENDELU Brno Tento projekt
Obecná genetika Mutace jako změna genetické informace a zdroj genetické variability Doc. RNDr. Ing. Eva PALÁTOVÁ, PhD. Ing. Roman LONGAUER, CSc. Ústav zakládání a pěstění lesů LDF MENDELU Brno Tento projekt
PREDIKTOR VLIVU AMINOKYSELINOVÝCH SUBSTITUCÍ NA FUNKCI PROTEINŮ
 VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA INFORMAČNÍCH TECHNOLOGIÍ ÚSTAV INFORMAČNÍCH SYSTÉMŮ FACULTY OF INFORMATION TECHNOLOGY DEPARTMENT OF INFORMATION SYSTEMS PREDIKTOR VLIVU
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA INFORMAČNÍCH TECHNOLOGIÍ ÚSTAV INFORMAČNÍCH SYSTÉMŮ FACULTY OF INFORMATION TECHNOLOGY DEPARTMENT OF INFORMATION SYSTEMS PREDIKTOR VLIVU
Genotypy absolutní frekvence relativní frekvence
 Genetika populací vychází z: Genetická data populace mohou být vyjádřena jako rekvence (četnosti) alel a genotypů. Každý gen má nejméně dvě alely (diploidní organizmy). Součet všech rekvencí alel v populaci
Genetika populací vychází z: Genetická data populace mohou být vyjádřena jako rekvence (četnosti) alel a genotypů. Každý gen má nejméně dvě alely (diploidní organizmy). Součet všech rekvencí alel v populaci
Využití internetových zdrojů při studiu mikroorganismů
 Využití internetových zdrojů při studiu mikroorganismů doc. RNDr. Milan Bartoš, Ph.D. bartosm@vfu.cz Přírodovědecká fakulta MU, 2012 Obsah přednášky 1) Práce se sekvenčními daty 2) Základní veřejně dostupné
Využití internetových zdrojů při studiu mikroorganismů doc. RNDr. Milan Bartoš, Ph.D. bartosm@vfu.cz Přírodovědecká fakulta MU, 2012 Obsah přednášky 1) Práce se sekvenčními daty 2) Základní veřejně dostupné
Genetická diverzita masného skotu v ČR
 Genetická diverzita masného skotu v ČR Mgr. Jan Říha Výzkumný ústav pro chov skotu, s.r.o. Ing. Irena Vrtková 26. listopadu 2009 Genetická diverzita skotu pojem diverzity Genom skotu 30 chromozomu, genetická
Genetická diverzita masného skotu v ČR Mgr. Jan Říha Výzkumný ústav pro chov skotu, s.r.o. Ing. Irena Vrtková 26. listopadu 2009 Genetická diverzita skotu pojem diverzity Genom skotu 30 chromozomu, genetická
Molekulární genetika
 Molekulární genetika Upozornění: ukončení semestru ZÁPOČTOVÝ TEST a) Dědičnost krevně skupinových systémů (AB0, MN, Rh) b) Přepis úseku DNA do sekvence aminokyselin c) Populační genetika výpočet frekvence
Molekulární genetika Upozornění: ukončení semestru ZÁPOČTOVÝ TEST a) Dědičnost krevně skupinových systémů (AB0, MN, Rh) b) Přepis úseku DNA do sekvence aminokyselin c) Populační genetika výpočet frekvence
Drift nejen v malých populacích (nebo při bottlenecku resp. efektu zakladatele)
") Drift nejen v malých populacích (nebo při bottlenecku resp. efektu zakladatele) Nově vzniklé mutace: nová mutace většinou v 1 kopii u 1 jedince mutace modelovány Poissonovým procesem Jaká je pravděpodobnost,
Drift nejen v malých populacích (nebo při bottlenecku resp. efektu zakladatele) Nově vzniklé mutace: nová mutace většinou v 1 kopii u 1 jedince mutace modelovány Poissonovým procesem Jaká je pravděpodobnost,
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/
 Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Genomika (KBB/GENOM) Poziční klonování Ing. Hana Šimková, CSc. Cíl přednášky - seznámení s metodou pozičního klonování genů
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Genomika (KBB/GENOM) Poziční klonování Ing. Hana Šimková, CSc. Cíl přednášky - seznámení s metodou pozičního klonování genů
Metabolismus aminokyselin. Vladimíra Kvasnicová
 Metabolismus aminokyselin Vladimíra Kvasnicová Aminokyseliny aminokyseliny přijímáme v potravě ve formě proteinů: důležitá forma organicky vázaného dusíku, který tak může být v těle využit k syntéze dalších
Metabolismus aminokyselin Vladimíra Kvasnicová Aminokyseliny aminokyseliny přijímáme v potravě ve formě proteinů: důležitá forma organicky vázaného dusíku, který tak může být v těle využit k syntéze dalších
Algoritmy a struktury neuropočítačů ASN P9 SVM Support vector machines Support vector networks (Algoritmus podpůrných vektorů)
") Algoritmy a struktury neuropočítačů ASN P9 SVM Support vector machines Support vector networks (Algoritmus podpůrných vektorů) Autor: Vladimir Vapnik Vapnik, V. The Nature of Statistical Learning Theory.
Algoritmy a struktury neuropočítačů ASN P9 SVM Support vector machines Support vector networks (Algoritmus podpůrných vektorů) Autor: Vladimir Vapnik Vapnik, V. The Nature of Statistical Learning Theory.
Metabolismus bílkovin. Václav Pelouch
 ZÁKLADY OBECNÉ A KLINICKÉ BIOCHEMIE 2004 Metabolismus bílkovin Václav Pelouch kapitola ve skriptech - 3.2 Výživa Vyvážená strava člověka musí obsahovat: cukry (50 55 %) tuky (30 %) bílkoviny (15 20 %)
ZÁKLADY OBECNÉ A KLINICKÉ BIOCHEMIE 2004 Metabolismus bílkovin Václav Pelouch kapitola ve skriptech - 3.2 Výživa Vyvážená strava člověka musí obsahovat: cukry (50 55 %) tuky (30 %) bílkoviny (15 20 %)
"Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Základy genetiky, základní pojmy
 "Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Základy genetiky, základní pojmy 1/75 Genetika = věda o dědičnosti Studuje biologickou informaci. Organizmy uchovávají,
"Učení nás bude více bavit aneb moderní výuka oboru lesnictví prostřednictvím ICT ". Základy genetiky, základní pojmy 1/75 Genetika = věda o dědičnosti Studuje biologickou informaci. Organizmy uchovávají,
Inovace studia molekulární a buněčné biologie
 Investice do rozvoje vzdělávání Inovace studia molekulární a buněčné biologie Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky. Investice do rozvoje vzdělávání
Investice do rozvoje vzdělávání Inovace studia molekulární a buněčné biologie Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky. Investice do rozvoje vzdělávání
Modelov an ı biologick ych syst em u Radek Pel anek
 Modelování biologických systémů Radek Pelánek Modelování v biologických vědách typický cíl: pomocí modelů se snažíme pochopit, jak biologické systémy fungují model zahrnuje naše chápání simulace ukazuje,
Modelování biologických systémů Radek Pelánek Modelování v biologických vědách typický cíl: pomocí modelů se snažíme pochopit, jak biologické systémy fungují model zahrnuje naše chápání simulace ukazuje,
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY
 VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA INFORMAČNÍCH TECHNOLOGIÍ ÚSTAV INFORMAČNÍCH SYSTÉMŮ FACULTY OF INFORMATION TECHNOLOGY DEPARTMENT OF INFORMATION SYSTEMS PREDIKCE ŠKODLIVOSTI
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA INFORMAČNÍCH TECHNOLOGIÍ ÚSTAV INFORMAČNÍCH SYSTÉMŮ FACULTY OF INFORMATION TECHNOLOGY DEPARTMENT OF INFORMATION SYSTEMS PREDIKCE ŠKODLIVOSTI
Emergence chování robotických agentů: neuroevoluce
 Emergence chování robotických agentů: neuroevoluce Petra Vidnerová, Stanislav Slušný, Roman Neruda Ústav Informatiky, AV ČR Kognice a umělý život VIII Praha 28. 5. 2008 Evoluční robotika: EA & neuronové
Emergence chování robotických agentů: neuroevoluce Petra Vidnerová, Stanislav Slušný, Roman Neruda Ústav Informatiky, AV ČR Kognice a umělý život VIII Praha 28. 5. 2008 Evoluční robotika: EA & neuronové
Určení molekulové hmotnosti: ESI a nanoesi
 Cvičení Určení molekulové hmotnosti: ESI a nanoesi ) 1)( ( ) ( H m z H m z M k j j j m z z zh M Molekula o hmotnosti M se nabije z-krát protonem, pík iontu ve spektru je na m z : ) ( H m z M z Pro dva
Cvičení Určení molekulové hmotnosti: ESI a nanoesi ) 1)( ( ) ( H m z H m z M k j j j m z z zh M Molekula o hmotnosti M se nabije z-krát protonem, pík iontu ve spektru je na m z : ) ( H m z M z Pro dva
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ
 VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA ELEKTROTECHNIKY A KOMUNIKAČNÍCH TECHNOLOGIÍ FACULTY OF ELECTRICAL ENGINEERING AND COMMUNICATION ÚSTAV BIOMEDICÍNSKÉHO INŽENÝRSTVÍ DEPARTMENT
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA ELEKTROTECHNIKY A KOMUNIKAČNÍCH TECHNOLOGIÍ FACULTY OF ELECTRICAL ENGINEERING AND COMMUNICATION ÚSTAV BIOMEDICÍNSKÉHO INŽENÝRSTVÍ DEPARTMENT
Metody studia historie populací. Metody studia historie populací. 1) Metody studiagenetickérozmanitosti komplexní fenotypové znaky, molekulární znaky.
 Metody studiagenetickérozmanitosti komplexní fenotypové znaky, molekulární znaky.") 1) Metody studiagenetickérozmanitosti komplexní fenotypové znaky, molekulární znaky. 2)Mechanizmy evoluce mutace, p írodnívýb r, genový posun a genový tok 3) Anagenezex kladogeneze-co je vlastn druh 4)Dva
1) Metody studiagenetickérozmanitosti komplexní fenotypové znaky, molekulární znaky. 2)Mechanizmy evoluce mutace, p írodnívýb r, genový posun a genový tok 3) Anagenezex kladogeneze-co je vlastn druh 4)Dva
MOLEKULÁRNÍ BIOLOGIE. 2. Polymerázová řetězová reakce (PCR)
") MOLEKULÁRNÍ BIOLOGIE 2. Polymerázová řetězová reakce (PCR) Náplň praktik 1. Izolace DNA z buněk bukální sliznice - izolační kit MACHEREY-NAGEL 2. PCR polymerázová řetězová reakce (templát gdna) 3. Restrikční
MOLEKULÁRNÍ BIOLOGIE 2. Polymerázová řetězová reakce (PCR) Náplň praktik 1. Izolace DNA z buněk bukální sliznice - izolační kit MACHEREY-NAGEL 2. PCR polymerázová řetězová reakce (templát gdna) 3. Restrikční
Molekulární základ dědičnosti
 Molekulární základ dědičnosti Dědičná informace je zakódována v deoxyribonukleové kyselině, která je uložena v jádře buňky v chromozómech. Zlomovým objevem pro další rozvoj molekulární genetiky bylo odhalení
Molekulární základ dědičnosti Dědičná informace je zakódována v deoxyribonukleové kyselině, která je uložena v jádře buňky v chromozómech. Zlomovým objevem pro další rozvoj molekulární genetiky bylo odhalení
Degenerace genetického kódu
 AJ: degeneracy x degeneration CJ: degenerace x degenerace Degenerace genetického kódu Genetický kód je degenerovaný, resp. redundantní, což znamená, že dva či více kodonů může kódovat jednu a tutéž aminokyselinu.
AJ: degeneracy x degeneration CJ: degenerace x degenerace Degenerace genetického kódu Genetický kód je degenerovaný, resp. redundantní, což znamená, že dva či více kodonů může kódovat jednu a tutéž aminokyselinu.
Jak měříme genetickou vzdálenost a co nám říká F ST
 Jak měříme genetickou vzdálenost a co nám říká F ST 1) Genetická vzdálenost a její stanovení Pomocí genetické rozmanitosti, kterou se populace liší, můžeme určit do jaké míry jsou si příbuznější jaká je
Jak měříme genetickou vzdálenost a co nám říká F ST 1) Genetická vzdálenost a její stanovení Pomocí genetické rozmanitosti, kterou se populace liší, můžeme určit do jaké míry jsou si příbuznější jaká je
Algoritmizace Dynamické programování. Jiří Vyskočil, Marko Genyg-Berezovskyj 2010
 Dynamické programování Jiří Vyskočil, Marko Genyg-Berezovskyj 2010 Rozděl a panuj (divide-and-conquer) Rozděl (Divide): Rozděl problém na několik podproblémů tak, aby tyto podproblémy odpovídaly původnímu
Dynamické programování Jiří Vyskočil, Marko Genyg-Berezovskyj 2010 Rozděl a panuj (divide-and-conquer) Rozděl (Divide): Rozděl problém na několik podproblémů tak, aby tyto podproblémy odpovídaly původnímu
Vyhledávání. doc. Mgr. Jiří Dvorský, Ph.D. Katedra informatiky Fakulta elektrotechniky a informatiky VŠB TU Ostrava. Prezentace ke dni 21.
 Vyhledávání doc. Mgr. Jiří Dvorský, Ph.D. Katedra informatiky Fakulta elektrotechniky a informatiky VŠB TU Ostrava Prezentace ke dni 21. září 2018 Jiří Dvorský (VŠB TUO) Vyhledávání 242 / 433 Osnova přednášky
Vyhledávání doc. Mgr. Jiří Dvorský, Ph.D. Katedra informatiky Fakulta elektrotechniky a informatiky VŠB TU Ostrava Prezentace ke dni 21. září 2018 Jiří Dvorský (VŠB TUO) Vyhledávání 242 / 433 Osnova přednášky
Aminokyseliny příručka pro učitele. Obecné informace: Téma otevírá kapitolu Bílkoviny, která svým rozsahem překračuje rámec jedné vyučovací hodiny.
 Obecné informace: Aminokyseliny příručka pro učitele Téma otevírá kapitolu Bílkoviny, která svým rozsahem překračuje rámec jedné vyučovací hodiny. Navazující učivo Před probráním tématu Aminokyseliny probereme
Obecné informace: Aminokyseliny příručka pro učitele Téma otevírá kapitolu Bílkoviny, která svým rozsahem překračuje rámec jedné vyučovací hodiny. Navazující učivo Před probráním tématu Aminokyseliny probereme
Strom života. Cíle. Stručná anotace
 Předmět: Doporučený ročník: Vazba na ŠVP: Biologie 1. ročník Úvod do taxonomie Cíle Studenti zařadí člověka do příslušných taxonů taxonomického systému. Studenti se seznámí s principem fylogenetického
Předmět: Doporučený ročník: Vazba na ŠVP: Biologie 1. ročník Úvod do taxonomie Cíle Studenti zařadí člověka do příslušných taxonů taxonomického systému. Studenti se seznámí s principem fylogenetického
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ VYHLEDÁVÁNÍ HOMOLOGNÍCH GENŮ DIPLOMOVÁ PRÁCE
 VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA ELEKTROTECHNIKY A KOMUNIKAČNÍCH TECHNOLOGIÍ ÚSTAV BIOMEDICÍNSKÉHO INŽENÝRSTVÍ FACULTY OF ELECTRICAL ENGINEERING AND COMMUNICATION DEPARTMENT
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA ELEKTROTECHNIKY A KOMUNIKAČNÍCH TECHNOLOGIÍ ÚSTAV BIOMEDICÍNSKÉHO INŽENÝRSTVÍ FACULTY OF ELECTRICAL ENGINEERING AND COMMUNICATION DEPARTMENT
Vztah genotyp fenotyp
 Evoluce fenotypu II Vztah genotyp fenotyp plán? počítačový program? knihovna? genotypová astrologie (Jablonka a Lamb) Modely RNA - různé vážení: A-U, G-C, G-U interakcí, penalizace za neodpovídající si
Evoluce fenotypu II Vztah genotyp fenotyp plán? počítačový program? knihovna? genotypová astrologie (Jablonka a Lamb) Modely RNA - různé vážení: A-U, G-C, G-U interakcí, penalizace za neodpovídající si
Molekulární základy dědičnosti. Ústřední dogma molekulární biologie Struktura DNA a RNA
 Molekulární základy dědičnosti Ústřední dogma molekulární biologie Struktura DNA a RNA Ústřední dogma molekulární genetiky - vztah mezi nukleovými kyselinami a proteiny proteosyntéza replikace DNA RNA
Molekulární základy dědičnosti Ústřední dogma molekulární biologie Struktura DNA a RNA Ústřední dogma molekulární genetiky - vztah mezi nukleovými kyselinami a proteiny proteosyntéza replikace DNA RNA
Teorie neutrální evoluce a molekulární hodiny
 Teorie neutrální evoluce a molekulární hodiny Teorie neutrální evoluce Konec 60. a začátek 70. let 20. stol. Ukazuje jak bude vypadat genetická variabilita v populaci a jaká bude rychlost evoluce v případě,
Teorie neutrální evoluce a molekulární hodiny Teorie neutrální evoluce Konec 60. a začátek 70. let 20. stol. Ukazuje jak bude vypadat genetická variabilita v populaci a jaká bude rychlost evoluce v případě,
analýzy dat v oboru Matematická biologie
 INSTITUT BIOSTATISTIKY A ANALÝZ Lékařská a Přírodovědecká fakulta, Masarykova univerzita Komplexní přístup k výuce analýzy dat v oboru Matematická biologie Tomáš Pavlík, Daniel Schwarz, Jiří Jarkovský,
INSTITUT BIOSTATISTIKY A ANALÝZ Lékařská a Přírodovědecká fakulta, Masarykova univerzita Komplexní přístup k výuce analýzy dat v oboru Matematická biologie Tomáš Pavlík, Daniel Schwarz, Jiří Jarkovský,
Pokročilé operace s obrazem
 Získávání a analýza obrazové informace Pokročilé operace s obrazem Biofyzikální ústav Lékařské fakulty Masarykovy univerzity Brno prezentace je součástí projektu FRVŠ č.2487/2011 (BFÚ LF MU) Získávání
Získávání a analýza obrazové informace Pokročilé operace s obrazem Biofyzikální ústav Lékařské fakulty Masarykovy univerzity Brno prezentace je součástí projektu FRVŠ č.2487/2011 (BFÚ LF MU) Získávání
NUKLEOVÉ KYSELINY. Základ života
 NUKLEOVÉ KYSELINY Základ života HISTORIE 1. H. Braconnot (30. léta 19. století) - Strassburg vinné kvasinky izolace matiére animale. 2. J.F. Meischer - experimenty z hnisem štěpení trypsinem odstředěním
NUKLEOVÉ KYSELINY Základ života HISTORIE 1. H. Braconnot (30. léta 19. století) - Strassburg vinné kvasinky izolace matiére animale. 2. J.F. Meischer - experimenty z hnisem štěpení trypsinem odstředěním
PODOBNOSTI PRIMÁRNÍ STRUKTURY
 VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA INFORMAČNÍCH TECHNOLOGIÍ ÚSTAV INFORMAČNÍCH SYSTÉMŮ FACULTY OF INFORMATION TECHNOLOGY DEPARTMENT OF INFORMATION SYSTEMS SHLUKOVÁNÍ PROTEINOVÝCH
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA INFORMAČNÍCH TECHNOLOGIÍ ÚSTAV INFORMAČNÍCH SYSTÉMŮ FACULTY OF INFORMATION TECHNOLOGY DEPARTMENT OF INFORMATION SYSTEMS SHLUKOVÁNÍ PROTEINOVÝCH
LEKCE 5 STATISTICKÁ INFERENCE ANEB ZOBECŇOVÁNÍ VÝSLEDKŮ Z VÝBĚROVÉHO NA ZÁKLADNÍ SOUBOR
 LEKCE 5 STATISTICKÁ INFERENCE ANEB ZOBECŇOVÁNÍ VÝSLEDKŮ Z VÝBĚROVÉHO NA ZÁKLADNÍ SOUBOR Ve většině případů pracujeme s výběrovým souborem a výběrové výsledky zobecňujeme na základní soubor. Smysluplné
LEKCE 5 STATISTICKÁ INFERENCE ANEB ZOBECŇOVÁNÍ VÝSLEDKŮ Z VÝBĚROVÉHO NA ZÁKLADNÍ SOUBOR Ve většině případů pracujeme s výběrovým souborem a výběrové výsledky zobecňujeme na základní soubor. Smysluplné
Hardy-Weinbergův zákon - cvičení
 Genetika a šlechtění lesních dřevin Hardy-Weinbergův zákon - cvičení Doc. Ing. RNDr. Eva Palátová, PhD. Ústav zakládání a pěstění lesů LDF MENDELU Brno Tento projekt je spolufinancován Evropským sociálním
Genetika a šlechtění lesních dřevin Hardy-Weinbergův zákon - cvičení Doc. Ing. RNDr. Eva Palátová, PhD. Ústav zakládání a pěstění lesů LDF MENDELU Brno Tento projekt je spolufinancován Evropským sociálním
Statistické řízení jakosti - regulace procesu měřením a srovnáváním
 Statistické řízení jakosti - regulace procesu měřením a srovnáváním Statistická regulace výrobního procesu (SPC) SPC = Statistical Process Control preventivní nástroj řízení jakosti, který na základě včasného
Statistické řízení jakosti - regulace procesu měřením a srovnáváním Statistická regulace výrobního procesu (SPC) SPC = Statistical Process Control preventivní nástroj řízení jakosti, který na základě včasného
 Evoluční genetika KBI/GENE Mgr. Zbyněk Houdek Evoluční teorie Evoluční teorii vyslovil Ch. Darwin v díle O původu druhů (1859), kde ukazoval, že druhy se postupně měnily v dlouhých časových periodách.
Evoluční genetika KBI/GENE Mgr. Zbyněk Houdek Evoluční teorie Evoluční teorii vyslovil Ch. Darwin v díle O původu druhů (1859), kde ukazoval, že druhy se postupně měnily v dlouhých časových periodách.
Úvod do optimalizace, metody hladké optimalizace
 Evropský sociální fond Investujeme do vaší budoucnosti Úvod do optimalizace, metody hladké optimalizace Matematika pro informatiky, FIT ČVUT Martin Holeňa, 13. týden LS 2010/2011 O čem to bude? Příklady
Evropský sociální fond Investujeme do vaší budoucnosti Úvod do optimalizace, metody hladké optimalizace Matematika pro informatiky, FIT ČVUT Martin Holeňa, 13. týden LS 2010/2011 O čem to bude? Příklady
Induktivní statistika. z-skóry pravděpodobnost
 Induktivní statistika z-skóry pravděpodobnost normální rozdělení Z-skóry umožňují najít a popsat pozici každé hodnoty v rámci rozdělení hodnot a také srovnávání hodnot pocházejících z měření na rozdílných
Induktivní statistika z-skóry pravděpodobnost normální rozdělení Z-skóry umožňují najít a popsat pozici každé hodnoty v rámci rozdělení hodnot a také srovnávání hodnot pocházejících z měření na rozdílných
Molekulárně biologické metody princip, popis, výstupy
 & Molekulárně biologické metody princip, popis, výstupy Klára Labská Evropský program pro mikrobiologii ve veřejném zdravotnictví (EUPHEM), ECDC, Stockholm NRL pro herpetické viry,centrum epidemiologie
& Molekulárně biologické metody princip, popis, výstupy Klára Labská Evropský program pro mikrobiologii ve veřejném zdravotnictví (EUPHEM), ECDC, Stockholm NRL pro herpetické viry,centrum epidemiologie
Struktura nukleových kyselin Vlastnosti genetického materiálu
 Struktura nukleových kyselin Vlastnosti genetického materiálu V předcházejících kapitolách bylo konstatováno, že geny jsou uloženy na chromozomech a kontrolují fenotypové vlastnosti a že chromozomy se
Struktura nukleových kyselin Vlastnosti genetického materiálu V předcházejících kapitolách bylo konstatováno, že geny jsou uloženy na chromozomech a kontrolují fenotypové vlastnosti a že chromozomy se
PREDIKCE VLIVU AMINOKYSELINOVÝCH MUTACÍ NA SEKUNDÁRNÍ STRUKTURU PROTEINŮ
 VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA INFORMAČNÍCH TECHNOLOGIÍ ÚSTAV INFORMAČNÍCH SYSTÉMŮ FACULTY OF INFORMATION TECHNOLOGY DEPARTMENT OF INFORMATION SYSTEMS PREDIKCE VLIVU
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA INFORMAČNÍCH TECHNOLOGIÍ ÚSTAV INFORMAČNÍCH SYSTÉMŮ FACULTY OF INFORMATION TECHNOLOGY DEPARTMENT OF INFORMATION SYSTEMS PREDIKCE VLIVU
Centrum aplikované genomiky, Ústav dědičných metabolických poruch, 1.LFUK
 ové technologie v analýze D A, R A a proteinů Stanislav Kmoch Centrum aplikované genomiky, Ústav dědičných metabolických poruch, 1.LFUK Motto : "The optimal health results from ensuring that the right
ové technologie v analýze D A, R A a proteinů Stanislav Kmoch Centrum aplikované genomiky, Ústav dědičných metabolických poruch, 1.LFUK Motto : "The optimal health results from ensuring that the right
Využití metagenomiky při hodnocení sanace chlorovaných ethylenů in situ Výsledky pilotních testů
 Využití metagenomiky při hodnocení sanace chlorovaných ethylenů in situ Výsledky pilotních testů Stavělová M.,* Macháčková J.*, Rídl J.,** Pačes J.** * Earth Tech CZ, s.r.o ** ÚMG AV ČR PROČ METAGENOMIKA?
Využití metagenomiky při hodnocení sanace chlorovaných ethylenů in situ Výsledky pilotních testů Stavělová M.,* Macháčková J.*, Rídl J.,** Pačes J.** * Earth Tech CZ, s.r.o ** ÚMG AV ČR PROČ METAGENOMIKA?
Teorie neutrální evoluce a molekulární hodiny
 Teorie neutrální evoluce a molekulární hodiny Teorie neutrální evoluce Konec 60. a začátek 70. let 20. stol. Ukazuje jak bude vypadat genetická variabilita v populaci a jaká bude rychlost divergence druhů
Teorie neutrální evoluce a molekulární hodiny Teorie neutrální evoluce Konec 60. a začátek 70. let 20. stol. Ukazuje jak bude vypadat genetická variabilita v populaci a jaká bude rychlost divergence druhů
Algoritmy pro shlukování prostorových dat
 Algoritmy pro shlukování prostorových dat Marta Žambochová Katedra matematiky a informatiky Fakulta sociálně ekonomická Univerzita J. E. Purkyně v Ústí nad Labem ROBUST 21. 26. leden 2018 Rybník - Hostouň
Algoritmy pro shlukování prostorových dat Marta Žambochová Katedra matematiky a informatiky Fakulta sociálně ekonomická Univerzita J. E. Purkyně v Ústí nad Labem ROBUST 21. 26. leden 2018 Rybník - Hostouň
ČVUT FEL X36PAA - Problémy a algoritmy. 4. úloha - Experimentální hodnocení algoritmů pro řešení problému batohu
 ČVUT FEL X36PAA - Problémy a algoritmy 4. úloha - Experimentální hodnocení algoritmů pro řešení problému batohu Jméno: Marek Handl Datum: 3. 2. 29 Cvičení: Pondělí 9: Zadání Prozkoumejte citlivost metod
ČVUT FEL X36PAA - Problémy a algoritmy 4. úloha - Experimentální hodnocení algoritmů pro řešení problému batohu Jméno: Marek Handl Datum: 3. 2. 29 Cvičení: Pondělí 9: Zadání Prozkoumejte citlivost metod
ve srovnání s eukaryoty (životnost v řádu hodin) u prokaryot kratší (životnost v řádu minut) na životnost / stabilitu molekuly mají vliv
 u prokaryot kratší (životnost v řádu minut) na životnost / stabilitu molekuly mají vliv") Urbanová Anna ve srovnání s eukaryoty (životnost v řádu hodin) u prokaryot kratší (životnost v řádu minut) na životnost / stabilitu molekuly mají vliv strukturní rysy mrna proces degradace každá mrna v
Urbanová Anna ve srovnání s eukaryoty (životnost v řádu hodin) u prokaryot kratší (životnost v řádu minut) na životnost / stabilitu molekuly mají vliv strukturní rysy mrna proces degradace každá mrna v
NGS analýza dat. kroužek, Alena Musilová
 NGS analýza dat kroužek, 16.12.2016 Alena Musilová Typy NGS experimentů Název Materiál Cílí na..? Cíl experimentu? amplikon DNA malý počet vybraných genů hledání variant exom DNA všechny geny hledání
NGS analýza dat kroužek, 16.12.2016 Alena Musilová Typy NGS experimentů Název Materiál Cílí na..? Cíl experimentu? amplikon DNA malý počet vybraných genů hledání variant exom DNA všechny geny hledání
Tomáš Oberhuber. Faculty of Nuclear Sciences and Physical Engineering Czech Technical University in Prague
 Tomáš Faculty of Nuclear Sciences and Physical Engineering Czech Technical University in Prague Buňka buňka je základní stavební prvek všech živých organismů byla objevena Robertem Hookem roku 1665 jednodušší
Tomáš Faculty of Nuclear Sciences and Physical Engineering Czech Technical University in Prague Buňka buňka je základní stavební prvek všech živých organismů byla objevena Robertem Hookem roku 1665 jednodušší
základní znaky živých systémů (definice života výčtem jeho vlastností) složitá organizace a řád regulace a udržování vnitřní homeostázy získávání a
 složitá organizace a řád regulace a udržování vnitřní homeostázy získávání a") definice života živý organismus je přirozeně se vyskytující sám sebe reprodukující systém, který vykonává řízené manipulace s hmotou, energií a informací základní znaky živých systémů (definice života
definice života živý organismus je přirozeně se vyskytující sám sebe reprodukující systém, který vykonává řízené manipulace s hmotou, energií a informací základní znaky živých systémů (definice života
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ
 VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA ELEKTROTECHNIKY A KOMUNIKAČNÍCH TECHNOLOGIÍ ÚSTAV BIOMEDICÍNSKÉHO INŽENÝRSTVÍ FACULTY OF ELECTRICAL ENGINEERING AND COMMUNICATION DEPARTMENT
VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ BRNO UNIVERSITY OF TECHNOLOGY FAKULTA ELEKTROTECHNIKY A KOMUNIKAČNÍCH TECHNOLOGIÍ ÚSTAV BIOMEDICÍNSKÉHO INŽENÝRSTVÍ FACULTY OF ELECTRICAL ENGINEERING AND COMMUNICATION DEPARTMENT
PROTEINY. Biochemický ústav LF MU (H.P.)
") PROTEINY Biochemický ústav LF MU 2013 - (H.P.) 1 proteiny peptidy aminokyseliny 2 Aminokyseliny 3 Charakteristika základní stavební jednotky proteinů geneticky kódované 20 základních aminokyselin 4 a-aminokyselina
PROTEINY Biochemický ústav LF MU 2013 - (H.P.) 1 proteiny peptidy aminokyseliny 2 Aminokyseliny 3 Charakteristika základní stavební jednotky proteinů geneticky kódované 20 základních aminokyselin 4 a-aminokyselina
Binární vyhledávací stromy pokročilé partie
 Binární vyhledávací stromy pokročilé partie KMI/ALS lekce Jan Konečný 30.9.204 Literatura Cormen Thomas H., Introduction to Algorithms, 2nd edition MIT Press, 200. ISBN 0-262-5396-8 6, 3, A Knuth Donald
Binární vyhledávací stromy pokročilé partie KMI/ALS lekce Jan Konečný 30.9.204 Literatura Cormen Thomas H., Introduction to Algorithms, 2nd edition MIT Press, 200. ISBN 0-262-5396-8 6, 3, A Knuth Donald
Malcomber S.T. (2000): Phylogeny of Gaertnera Lam. (Rubiaceae) based on multiple DNA markers: evidence of a rapid radiation in a widespread,
: Phylogeny of Gaertnera Lam. (Rubiaceae) based on multiple DNA markers: evidence of a rapid radiation in a widespread,") Malcomber S.T. (2000): Phylogeny of Gaertnera Lam. (Rubiaceae) based on multiple DNA markers: evidence of a rapid radiation in a widespread, morphologically diverse genus. Evolution 56(1):42-57 Proč to
Malcomber S.T. (2000): Phylogeny of Gaertnera Lam. (Rubiaceae) based on multiple DNA markers: evidence of a rapid radiation in a widespread, morphologically diverse genus. Evolution 56(1):42-57 Proč to