Molecular-cytogenetic analysis of chromosome 11 aberrations in hematological malignancies
|
|
|
- Marian Král
- před 8 lety
- Počet zobrazení:
Transkript

1 Univerzita Karlova v Praze 1. lékařská fakulta Doktorské studijní programy v biomedicíně Studijní obor: Molekulární a buněčná biologie, genetika a virologie RNDr. Iveta Šárová Molekulárně cytogenetická analýza aberací chromosomu 11 u hematologických malignit Molecular-cytogenetic analysis of chromosome 11 aberrations in hematological malignancies Disertační práce Vedoucí práce: RNDr. Jana Březinová, Ph.D. Praha 2012
2 Prohlášení: Prohlašuji, že jsem závěrečnou práci zpracovala samostatně a že jsem řádně uvedla a citovala všechny použité prameny a literaturu. Současně prohlašuji, že práce nebyla využita k získání jiného nebo stejného titulu Souhlasím s trvalým uložením elektronické verze mé práce v databázi systému meziuniverzitního projektu Theses.cz za účelem soustavné kontroly podobnosti kvalifikačních prací. V Praze dne.. Iveta Šárová 2
3 Identifikační záznam: ŠÁROVÁ, Iveta. Molekulárně cytogenetická analýza aberací chromosomu 11 u hematologických malignit. [Molecular-cytogenetic analysis of chromosome 11 aberrations in hematological malignancies]. Praha, s., 7 příl. Disertační práce (Ph.D.). Ústav hematologie a krevní transfúze/univerzita Karlova v Praze, 1. lékařská fakulta, Centrum nádorové cytogenetiky ÚLBLD. Vedoucí závěrečné práce Březinová, Jana. 3
4 Abstrakt Změny chromosomu 11 patří mezi časté cytogenetické nálezy u hematologických malignit. Zlomové místo se obvykle nachází v oblasti 11q23.3, v proto-onkogenu MLL. Vzácně se zlomy vyskytují i v jiných oblastech, což svědčí o účasti i dalších genů. V naší studii jsme se zaměřili na identifikaci zlomů a oblastí amplifikace/delece na chromosomu 11 v buňkách kostní dřeně/periferní krve nemocných s akutní myeloidní leukémií (AML). Na chromosomu 11 jsme identifikovali množství rekurentních i náhodných zlomových míst (rekurentní v genech MLL (11q23.3) a NUP98 (11p15.4) a v oblastech 11p13, 11p12 a 11q13.2) a vyhodnotili jsme deletované a duplikované/amplifikované oblasti. Poukázali jsme na nové kandidátní geny s možnou úlohou v patogenezi AML. Oproti přestavbám MLL genu, jsme prokázali spojitost ostatních změn chromosomu 11 se starším věkem nemocných, komplexním karyotypem, nebalancovanou změnou a krátkou dobou přežití. FISH skríningové vyšetření se ukázalo velmi přínosné u pacientů s nedostatkem dělících se buněk a pro detekci kryptických přestaveb MLL genu. Studium chromosomových změn slouží nejen ke klinické stratifikaci nemocných do prognostických skupin, ale je i nezbytným podkladem pro identifikaci genů, které asociují se vznikem a progresí nádorů. Analýzy těchto genů a jejich produktů nám pomáhají porozumět patogenezi maligní transformace. Významně přispívají k vývoji nových terapeutik a cílené léčby. Klíčová slova: akutní myeloidní leukémie, MLL, komplexní karyotyp, FISH mapování. 4
5 Abstract Chromosome 11 abnormalities are found in many hematological malignancies. In acute myeloid leukemia (AML), a proto-oncogene MLL (11q23.3) is frequently altered. However, rearrangements to other regions of chromosome 11 have been reported. Therefore, we have identified and characterized the chromosome 11 breakpoints and common deleted and amplified areas in the bone marrow or peripheral blood cells of newly diagnosed patients with AML. Many recurrent and random chromosome 11 breakpoints were identified (recurrent in bands 11p15.4 (in NUP98 gene), 11q23.3 (in the MLL gene), 11p13, 11p12 and 11q13.2) and deleted or duplicated/amplified regions were determined. We notified new possibly significant genes in the development of AML. Contrary to the MLL rearrangements, patients with other chromosome 11 changes were older, with complex karyotype, unbalanced aberrations and short survival. FISH screening was proved very helpful in case of deviding cells lack and cryptic MLL gene rearrangement. In conclusion, molecular analyses of chromosomal breakpoints and amplified or deleted areas are very important not only for the patient stratification into specific prognostic and clinical subgroups but also for the identification of genes involved in tumour pathogenesis. Further investigation of the affected genes and their protein products will improve our understanding of the oncogenesis of AML and could be clinically applied for the designation of more effective therapeutic approach. Key words: acute myeloid leukemia (AML), MLL, complex karyotype, FISH mapping. 5
6 Na tomto místě bych velmi ráda vyjádřila své poděkování předvším RNDr. Janě Březinové, Ph.D za odborné vedení práce, poskytnutí řady konzultací ke zvolené problematice a trpělivosti. Také za podporu a nezdolný optismus. Velmi ráda bych také poděkovala Prof. Ing. K. Michalové za srdečné přijetí do kolektivu profesionálních cytogenetiků a poskytnutí možnosti vypracovat si tuto práci na velice dobře vybaveném, akreditovaném pracovišti. Prof. Ing. K. Michalové a doc. Z. Zemanové děkuji za mnoho užitečných rad a konzultací, a to především při získávání zkušeností v oblasti publikačních dovedností a prezentace výsledků. Poděkovat musím i celému zbylému kolektivu z Centra nádorové cytogenetiky ÚLBLD VFN a 1.LF UK a především kolegyním z Cytogenetické laboratoře ÚHKT za velmi přátelské prostředí. Za rychlé zpracování statistických dat a milý přístup děkuji i Ing. A. Dohnalové a také všem klinickým lékařům, kteří nám věnovali čas a poskytli klinická data nemocných nutná pro statistické zpracování. V neposlední řadě musím poděkovat své rodině nejen za vytvoření příznivých studijních podmínek, ale také za nezbytnou podporu a pochopení. 6
7 SEZNAM ZKRATEK AF AML ATM BCR CCND1 DCDC del der FAB FISH GAL ins inv lncrna MAML2 mband MDS mfish MLL MLLT mrna MTL5 NUP98 p PTD q RARA t TKD TP53 WHO ALL1 fused gene from chromosome akutní myeloidní leukemie ataxia telangiectasia mutated breakpoint cluster region cyclin D1 doublecortin domain-containing protein delece derivovaný chromosom French-American-British fluorescenční in situ hybridizace galanin inzerce inverze long non-coding RNA mastermind-like protein 2 mnohobarevné pruhování myelodysplastický syndrom mnohobarevná fluorescenční in situ hybridizace myeloid/lymphoid leukemia myeloid/lymphoid leukemia translocated to, microrna metallothionein-like 5, testis-specific (tesmin) nucleoporin 98 KDa označení pro krátké rameno chromosomu parciální tandémová duplikace označení pro dlouhé rameno chromosomu retinoic acid receptor, alpha translokace transplantace kostní dřeně gen pro protein 53 kda World Health Organization 7
8 OBSAH 1. ÚVOD CÍLE PRÁCE PŘEHLED LITERATURY Chromosom Molekulární podstata nádorových onemocnění Změny chromosomu 11 u hematologických malignit Změny dlouhého ramene chromosomu 11 (11q) Změny krátkého ramene chromosomu 11 (11p) Akutní myeloidní leukémie (AML) Chromosomové změny u AML a jejich prognostický význam Molekulární markery u AML a jejich prognostický význam AML u dětí MATERIÁL A METODY Materiál Soubor pacientů Metodika Klasická cytogenetická analýza Molekulární cytogenetická analýza Molekulárně genetická analýza Statistická analýza VÝSLEDKY Změny chromosomu 11 u dospělých s AML Mapování zlomů Charakteristika a klinický význam zlomových míst Změny chromosomu 11 u dětí s AML Skríningové vyšetření u AML DISKUZE ZÁVĚR SEZNAM CITOVANÉ LITERATURY SEZNAM PŘÍLOH SEZNAM PUBLIKACÍ, PŘEDNÁŠEK A POSTEROVÝCH SDĚLENÍ...102
9 1. ÚVOD Chromosomové a genetické změny jsou nezbytným zdrojem pro vznik a vývoj eukaryotních organismů v evolučním měřítku. Právě tyto změny vedly k vytvoření a variabilitě všech rostliných a živočišných druhů, tak jak je známe dnes. Je paradoxem, že tyto mechanizmy nemusí vést jen k zrodu, ale i zániku, a to pokud k nim dochází na buněčné úrovni v rámci jedince. Změny v DNA sekvenci, její struktuře nebo epigenetické regulaci umožňují buňce získat selekční výhodu nad ostatními a spustit tak klonální expanzi označovanou jako nádor. Hypotéza o chromosomových abnormalitách jako zdrojích maligních onemocnění byla již formulována v roce 1914 německým biologem Theodorem Boverim a následně potvrzena řadou dalších studií. Objevení Filadelfského chromosomu, výsledku translokace t(9;22)(q34;q11), u chronické myeloidní leukémie v roce 1960 spustilo vlnu cytogenetických studií hledajících podobné změny i u dalších hematologických a solidních nádorů a způsobilo rozvoj a expanzi molekulárně cytogenetických metod, které aberace snadněji zachytily a charakterizovaly. V současné době bylo u nádorových onemocnění popsáno přes patologických karyotypů. Jejich archivace si vynutila vznik řady internetových databází jako je například Mitelman Database of Chromosome Aberrations in Cancer ( nebo Atlas of Genetics and Cytogenetics in Oncology and Hematology ( Statistické studie skupin nemocných s hematologickou malignitou a se stejnou chromosomovou změnou dokázaly, že cytogenetická analýza je nedílnou součástí vyšetřovacích metod. Stala se velmi důležitým klinickým nástrojem, který při diagnostickém odběru přispívá k upřesnění diagnózy, určení stádia nemoci, stanovení prognózy a následnému výběru léčebné terapie. Mnoho studií již prokázalo, že přítomnost cytogeneticky abnormálního klonu je nepříznivým prognostickým znakem jak u hematologických, tak i solidních nádorů. O míře nepříznivosti rozhoduje i míra komplexity chromosomových změn. Informativní význam cytogenetické analýzy neklesá ani v průběhu onemocnění, kdy umožňuje sledovat jeho progresi, případně transformaci, ukazuje na úspěšnost léčby po chemoterapii nebo transplantaci kostní dřeně a v neposlední řadě může být použita i k stanovení minimální reziduální choroby. Kromě klinického významu přinesla cytogenetická analýza i mnoho velmi důležitých poznatků ve studiu onkogeneze a zůstává významným nástrojem pro 9
10 lokalizaci konkrétních alterovaných genů, především je-li změna balancovaná a tím je pro řadu jiných celogenomových metod jako je například array analýza nedetekovatelná. Molekulárním genetikům poskytuje velmi cenné informace o tom, které místo je vhodné sekvenovat a identifikovat tak postižené geny. Následné biologické a funkční analýzy těchto genů jsou předpokladem pro vývoj individuální a cílené terapie. Příkladem významu objasnění molekulární podstaty chromosomových změn je translokace t(15;17)(q22;q21), charakteristická pro aktutní promyelocytární leukémii, při níž dochází k přestavbě receptoru kyseliny retinové. Zavedení cílené léčby pomocí inhibitorů této kyseliny vedlo k přehodnocení dřívě prognosticky nepříznivého onemocnění do skupiny prognosticky velmi příznivé s dlouhodobým stavem remise. V naší studii jsme se zaměřili na detekci a charakteristiku změn na chromosomu 11, protože patří mezi časté cytogenetické nálezy u hematologických malignit. Většina cytogenetických i genetických analýz se zabývá oblastí 11q23.3, kde je lokalizovaný protoonkogen MLL. Mnohé ostatní změny chromosomu 11 se vyskytují vzácně a zůstávají tak cytogeneticky i geneticky málo probádané. Studií jsme tyto abnormality lépe definovali a charakterizovali jak na základě cytogenetickém, tak i klinickém. 10
11 2. CÍLE PRÁCE Aberace chromosomu 11 byly popsány u řady hematologických malignit, nejčastěji u akutní myeloidní leukémie (AML). Proto jsme se v naší studii zaměřili na detekci aberací chromosomu 11 právě u tohoto onemocnění. Cílem studie bylo: charakterizovat změny chromosomu 11 u dospělých a dětských pacientů s nově diagnostikovanou AML na molekulárně-cytogenetické úrovni určit a mapovat rekurentní zlomová místa a oblasti amplifikace/delece na chromosomu 11 pro identifikaci možných genů asociovaných s leukémií zjistit, zda se zlomová místa a frekvence jejich výskytu liší v reciprokých, nereciprokých a komplexních přestavbách, u primárních a sekundárních onemocnění, u dětí a dospělých vyhodnotit vztah mezi zlomovým místem a dalšími klinickými ukazateli, posoudit význam odlišných zlomových míst pro prognózu nemocných zhodnotit význam skríningového molekulárně-cytogenetického vyšetření zaměřeného na nejčastější chromosomové změny u AML (abnormality oblasti 11q23.3, delece dlouhých ramen chromosomů 5 a 7 a početní změny chromosomů 8 a 9) 11
12 3. PŘEHLED LITERATURY 3.1. Chromosom 11 Dle ISCN (International System of Human Cytogenetic Nomenclature) nomenklatury patří chromosom 11 do skupiny C, představující středně velké, submetacentrické chromosomy (Shaffer a kol. 2009). Představuje přibližně 4 až 4,5 % lidského genomu (asi 134,5 Mb včetně centromery), čímž se řadí mezi nejvíce genově bohaté chromosomy v lidském genomu. Euchromatin tvoří až 99,8 % chromosomu (Taylor a kol. 2006) Na chromosomu 11 bylo dosud identifikováno protein kódujích genů z celkového množství až (viz tabulka 1). V průměru se odhaduje přítomnost 11,6 genů na 1 Mb. Jedna třetina protein kódujících genů se překrývá. Zajímavostí je přítomnost značného množství CpG ostrůvků a sestřihových variant genů. Taylor a kol. (2006) nalezli nejméně 2 různé sestřihové varianty u 805 (52,8 %) genů lokalizovaných na chromosomu 11. Celkem bylo popsáno variant pro exprimovaných genů. Na chromosomu 11 se nachází řada významných genů i tzv. genových rodin. Z 856 genů nesoucích informaci pro čichové receptory se více než 40 % z nich vyskytuje právě na tomto chromosomu (Taylor a kol. 2006). Chromosom 11 má velmi významnou roli v historii molekulární genetiky, a to díky genu pro ß-řetězec hemoglobinu, který se nachází na jeho krátkém rameni v pruhu 11p15.5. Jedná se o jeden z nejlépe prostudovaných genů vůbec. Mutace, při níž dochází v 6. pozici k záměně kyseliny glutamové za valin, vede k změně tvaru červených krvinek z tzv. promáčknutých piškotů na protažené srpky. Onemocnění je známé jako srpkovitá anémie. Spojitost genetické nemoci s mutací specifického proteinu, kterou vyslovil slavný americký fyzikální chemik Linus Pauling v roce 1949, se stala historickým milníkem v oboru molekulární biologie. V současné době jsou změny chromosomu 11 pokládány za podstatu více než 100 vrozených genetických onemocnění. Příkladem jsou Beckwith-Wiedemannův syndrom, Emanuelův syndrom, Jacobsenův syndrom, neuroblastom, Russell-Silverův syndrom, Wilmsův tumor a další. Sledována byla spojitost i s různými typy maligních onemocnění (Nowak a Shows, 1995). 12
13 Tabulka 1. Výsledky sekvenace chromosomu 11 (převzato z Taylor a kol. 2006) Molekulární podstata nádorových onemocnění Vznik, vývoj a progrese nádorového onemocnění jsou založeny na kumulaci genetických mutací a epigenetických změn odehrávajících se v jediné somatické buňce. U hematologických malignit lze mnohé genetické změny sledovat již na cytogenetické úrovni. Některé chromosomové aberace se vyskytují pouze náhodně a nemají tak diagnostické, prognostické ani terapeutické uplatnění. Důležitou roli hrají nenáhodné chromosomové abnormality, které se velmi specificky vyskytují u konkrétního typu neoplázie a mají značný klinický význam (Mitelman 2010). Genetické a epigenetické změny v buňce vedou k alteraci normálních buněčných procesů jako je buněčný růst, proliferace a diferenciace (Hanahan a Weinberg 2011). Ve výsledku dochází k nekontrolovatelnému dělení této buňky a založení nádorového klonu (nález nejméně dvou mitos se stejnou chromosomovou přestavbou nebo se stejným nadpočetným chromosomem, anebo výskyt tří mitos se stejným chybějícím chromosomem (Shaffer a kol. 2009). Při nádorové progresi lze sledovat klonální vývoj nemoci, a to přítomností dalších genetických abnormalit označovaných jako sekundární. Obvykle vedou k zhoršení prognózy nemocného. U nádorových onemocnění se setkáváme s těmito chromosomovými abnormalitami: nebalancované ztráty/zmnožení genetického materiálu nebo balancované chromosomové aberace (Sandberg a Meloni-Ehrig 2010). 13
14 Z cytogenetického hlediska lze ztrátu genetického materiálu vyjádřit jako deleci nebo monosomii. Postiženy bývají oblasti obsahující nádorové supresorové geny nebo mutátorové geny, účastnící se reparace DNA. Produkty nádorových supresorových genů inhibují buněčný cyklus, čímž brání buňce v nekontrolovatelné proliferaci nutné k vzniku nádoru. Zmnožení genetického materiálu můžeme pozorovat ve formě amplifikace, duplikace, trisomie nebo polyploidie. Patologický efekt spočívá ve zvýšení genové dávky vedoucí k nadprodukci příslušných genových produktů. Amplifikované kopie genu na chromosomu vytváří homogenně se barvící oblast (HSRs). Mohou se ale vyskytovat i samostatně jako množství acentrických fragmentů tzv. double minutes (DMs). Amplifikace ovlivňují především protoonkogeny, jejichž produkty pozitivně regulují buněčný cyklus a diferenciaci. Mezi chromosomové aberace bez ztráty nebo zisku genetického materiálu patří reciproké translokace, inverze a inzerce. Postiženy bývají jak protoonkogeny, nádorové supresorové geny, tak i mutátorové geny. Změny v uspořádání genetického materiálu vyvolávají dva zásadní typy změn: deregulaci transkripce postiženého genu nebo jeho strukturní přestavby - syntézu změněného produktu s odlišnou funkcí. V poslední době se studium vzniku nádorů upírá také na mikrorna jako na důležitého zprostředkovatele iniciace a progrese nádorů. Cílem těchto krátkých regulačních RNA jsou protoonkogeny nebo nádorové supresorové geny. Exprese mikrorna je ovlivňována genetickými a epigenetickými změnami zahrnujícími delece, amplifikace, bodové mutace a DNA metylace (Cho 2007). MikroRNA se jeví jako slibný diagnostický a patrně i prognostický molekulární ukazatel nádorových onemocnění Změny chromosomu 11 u hematologických malignit Jak již bylo uvedeno, abnormality chromosomu 11 byly popsány u řady maligních onemocnění, predevším u leukémií a lymfomů. S relativně častým nálezem aberací chromosomu 11 se setkáváme u akutní myeloidní leukémie (AML), myelodysplatického syndromu (MDS), akutní lymfatické leukémie (ALL), chronické lymfatické leukémie (CLL) a mnohočetného myelomu (MM). Postižena bývají především dlouhá ramena chromosomu
15 Změny dlouhého ramene chromosomu 11 (11q) Nejčastější abnormality chromosomu 11 u hematologických malignit souvisí s oblastí 11q23, která zahrnuje 10 až 15 Mb. Výzkum u sekundárních leukémií prokázal, že přestavby 11q23 jsou stejně frekventované jak u de novo, tak i sekundárních leukémií, způsobených léčbou zvláště pomocí inhibitorů topoizomerázy II, alkylačních činidel či radiace (Bloomfield a kol. 1998). Molekulárními analýzami zlomových míst v pruhu 11q23 došlo k identifikaci genu MLL (Zemin-van der Poel a kol. 1991). Oblast je však příliš velká na to, aby se její přestavby vždy týkaly tohoto genu. Mezi další často postižené geny u hematologických malignit patří i CCND1 (11q13) a ATM (11q22), vzácně byly sledovány abnormality těchto oblastí: 11q22-q23 (gen DDX10), 11q14.1 (gen GAB2), 11q24.1 (gen MIR100), 11q13 (gen NUMA1) Přehled dalších genů lokalizovaných na 11q je uveden v tabulce 2. Tabulka 2. Alterované geny na 11q u hematologických malignit. Oblast Gen Funkce Změna Onemocnění Poznámka 11q11 MACROD1 signální transdukce t(11;21)(q13;q22) myeloidní leukémie, MDS 11q13 RELA signální transdukce mutace lymfoproliferace 11q13 MYEOV neznámá t(11;14)(q13;q32) MM exprese 11q13 CCND1 regulátor buněčného t(11;14)(q13;q32) B-buněčné leukémie, MM exprese cyklu 11q13 NUMA1 poloha dělícího vřeténka t(11;17)(q13;q21) AML-M3 11q13 INPPL1 signální transdukce exprese CML 11q14.1 GAB2 signální transdukce amplifikace myeloidní leukémie 11q14 PICALM signální transdukce t(10;11)(p13;q14) T-buněčné ALL 11q21 CEP57 tvorba cytoskeletu delece MDS 11q21 MAML2 signální transdukce inv(11)(q21q23) sekundární leukémie, MDS 11q22 BIRC3 inhibitor apoptózy t(11;18)(q21;q21) lymfom 11q22 ATM signální transdukce delece CLL 11q22 DDX10 RNA helikáza inv(11)(p15q22) AML, MDS 11q23.1 ARHGAP20 signální transdukce mutace B-CLL 11q23.1 POU2AF1 transkripční koaktivátor amplifikace MM, lymfom 11q23.1 ZBTB16 transkripční faktor t(11;17)(q23;q21) AML-M3 11q23.3 MLL transkripční faktor translokace AML, ALL, MDS 11q23.3 ARHGEF12 signální transdukce del(11)(q23q23) AML fúzní gen 11q24.1 MIR100 regulační RNA změna exprese AML, ALL 11q24.2 SPA17 signální transdukce exprese MM možná chemorezistence Vysvětlivky: MDS (myelodysplastický syndrom), MM (mnohočetný myelom), AML-M3 (akutní myeloidní leukémie podtyp M3), CML (chronická lymfatická leukémie), ALL (akutní lymfatická leukémie), CLL (chronická lymfatická leukémie). 15
16 MLL gen (myeloid/lymphoid or mixed lineage leukemia) Aberace MLL genu jsou popisovány u 10 % všech akutních hematologických malignit zahrnujících myeloidní, lymfatické, bifenotypické, sekundární a dětské leukémie (Chowdhury a Brady 2008, Qian a kol. 2010). Protoonkogen MLL obsahuje 37 exonů a velikostně přesahuje 100 kb. Transkripce probíhá ve směru od centromery. Gen kóduje protein obsahující aminokyselin odpovídající 431 kda. Posttranslačně je sestřižen specifickou taspázou (treoninendopeptidázou) do většího 320 kda N-koncového a menšího 180 kda C- koncového fragmentu (viz obrázek 1). Obě části zůstávají nadále nekovalentně vázány ve formě dimeru. MLL protein se nachází v jádře, kde je součástí komplexu tvořeného nejméně 30 proteiny. Jeho role spočívá v remodelaci, acetylaci, deacetylaci a metylaci nukleosomů a histonů. Ovlivňuje buněčnou diferenciaci, apoptózu, proliferaci a regulaci buněčného cyklu (Hess 2004, Marschalek 2011). Obrázek 1. Schématické znázornění MLL proteinu. Vysvětlivky: AT hooks (AT-vazebné motivy), SNL (sekvence nukleární lokalizace), RD (represní doména, cystein bohatý region homologní k savčí DNA - metyltransferáze), PHD+bromo (motivy zinkových prstů a bromodoména; protein - protein interakce), FYRN/FYRC (N- a C-koncová část proteinu), TAD (transaktivační doména; váže CBP (CREB vazebný protein), acetylace histonů H3 a H4 v oblasti HOX genů), SET (serin a treonin bohátá doména; homodimerizace; metylace histonů H3 v regionu HOX genů), MBR (hlavní zlomový region), HDAC (histondeacetyláza). Převzato z k datu Velmi důležitou funkci má MLL protein během embryogeneze, kdy reguluje transkripci genů homeoboxu (HOX) (Slany 2005). Neiniciuje jejich expresi, ale udržuje ji po čas raného skeletárního, kraniofaciálního, neurálního a hematopoetického vývoje. V dospělosti je vyžadován pro diferenciaci myeloidních buněk. Uplatňuje se při proliferaci a přežití či nepřežití multipotentních progenitorů (Guenther a kol. 2005). Předpokládá se, že hlavním mechanizmem leukemické transformace iniciované MLL onkogenem je právě deregulace specifických HOX genů. Teorii potvrzuje vyšší exprese 16
17 HOXA7, HOXA9 a homeoboxového kofaktoru MEIS1 u leukémií s MLL abnormalitami (Slany 2005, Marschalek a kol. 2010). Zlomové místo v genu je obvykle lokalizováno uvnitř 8,3 kb regionu, označovaného BCR (breakpoint cluster region), mezi 5. a 11. exonem (dle nomenklatury NM_ exony 7 až 13) (Gu a kol. 1992). Umístění a frekvenci zlomových míst ovlivňuje ve velké míře chromatinová struktura, přítomnost repetitivních sekvencí a místa pro vazbu topoizomerázy II (Gu a kol. 1994, Strissel a kol. 1998). Přibližně polovina MLL přestaveb souvisí s intronem 6, kde je nejvyšší incidence Alu repetitivních sekvencí (proximální část BCR). U sekundární formy leukémie se zlom obvykle nachází v distální části BCR, kde je lokalizováno vazebné místo pro topoizomerázu II. Vysvětluje to vznik leukémií způsobených léčbou inhibitory tlumícími ligázovou funkci enzymu, který poté zanechává volné konce DNA. To může vést k nehomologní rekombinaci mezi MLL genem a partnerským genem (Braekeleer 2005, Aplan 2006, Zhang a Rowley 2006). Změnami MLL genu mohou být translokace, amplifikace, duplikace a delece, zřídka se objevují inverze nebo inzerce. Translokace a amplifikace se vyskytují nejčastěji. Přestavba MLL genu vede k vzniku fúzního genu tj. genu vzniklého splynutím 5 konce MLL genu s 3 koncem partnerského genu (Rowley a kol. 1992, Daser a Rabbitts 2004). Výsledný fúzní gen obsahující 5 MLL je obvykle lokalizovaný na derivovaném chromosomu 11. V případě transkripčně opačně orientovaného partnerského genu než je gen MLL je k vzniku fúzního genu zapotřebí dalších mechanizmů jako je inverze nebo inzerce (Daser a Rabbitts 2004, Braekeleer 2005, Douet-Guilbert a kol. 2005). Obvykle mají MLL translokace reciprokou, balancovanou povahu. Na nukleotidové úrovni však mohou být přítomny i krátké delece, inverze, inzerce nebo amplifikace (Aplan 2006). Dle databáze Atlas of Genetics and Cytogenetics in Oncology and Hematology bylo do roku 2010 popsáno 85 různých chromosomových partnerských oblastí, z nichž 66 bylo již charakterizováno i na molekulární úrovni (viz obrázek 2). Nejčastěji se vyskytují tyto translokace: t(6;11)(q27;q23), t(9;11)(p21;q23), t(10;11)(p12;q23) a t(11;19)(q23;p13) u AML a t(4;11)(q21;q23) a t(9;11)(p21;q23) u ALL (Gue a kol. 2006). Obecně lze fúzní partnery MLL genu rozdělit do 2 skupin: jaderné proteiny s transkripční aktivitou (př. AFF1 (AF4), MLLT3 (AF9), MLLT10 (AF10), ENL 17

18 (MLLT1), ELL) a proteiny cytoplazmatické (př. MLLT4 (AF6), SEPT2, SEPT4, SEPT6, SEPT9, SEPT11). Existuje několik hypotéz o funkci chimérického proteinu. Může neutralizovat normální funkci MLL proteinu kompeticí o cílová vazebná místa nebo vytvořit nový transkripční faktor fúzí DNA vazebného motivu s transaktivační doménou svého partnera. Dalším mechanizmem leukemogeneze je oligomerizace MLL genu, způsobená přítomností dimerizační domény partnerského proteinu (Daser a Rabbitts 2004, So a Cleary 2004, Dou a Hess 2008). Současný pohled podporuje teorii nabytí nových onkogenních vlastností vlivem fúze s partnerským genem. To je v souladu s faktem, že heterozygotní ztráta MLL genu nevede u myší k vzniku leukémie (Yu a kol. 1995, Liu a kol. 2007, Takáčová a kol. 2007). Obrázek 2. MLL gen a jeho fúzní partnerské geny. Převzato z k datu Odlišnou alterací MLL genu jsou parciální tandémové duplikace (PTD). Ty jsou popisovány u 6 10 % dospělých nemocných s AML převážně s normálním karyotypem nebo trisomií chromosomu 11 jako samostatnou abnormalitou (Caligiuri a kol. 1996, Caligiuri a kol. 1998, Whitman a kol. 2001, Döhner a kol. 2002, Mrozek a 18
19 kol. 2007). Duplikace MLL genu obvykle zahrnuje dle NM_ exony 2 až 8 (Caligiuri a kol. 1996). V proteinu se PTD projeví duplikací N-konce. MLL PTD byly pozorovány i u zdravé populace avšak s menší frekvencí a nezahrnující typické exony (Basecke a kol. 2006). Velmi vzácně může dojít k duplikaci již translokovaného MLL genu. Takto vzniklé parciální duplikace, obsahující část DNA z jiného chromosomu, se označují jako netandemové (PNTD). Tyto aberace nejsou cytogeneticky detekovatelné a jejich identifikace vyžaduje více citlivé metody jako je například reverzní transkripční polymerázová řetězcová reakce (RT-PCR). CCND1 (cyclin D1) Gen CCND1, kódující cyklin D1, o velikosti 4,5 kb obsahuje 5 exonů. Protein je tvořený 295 aminokyselinami celkově o 36 kda. Nachází se v jádře, kde hraje důležitou roli v regulaci buněčného cyklu, a to v přechodu buňky z G1 do S fáze. Jeho exprese je závislá na fázi buněčného cyklu, v které se buňka nachází. Maximálně se exprimuje v G1 fázi a nejméně v S fázi cyklu. Mutace cyklinu D1 byly nalezeny u nejrůznějších typů neoplázií, z leukémií jsou přítomny u B-buněčných leukémií a lymfomů, méně u myeloidních leukémií (Jaroslav a kol. 2005). Mezi nejvýznamnější přestavby patří translokace t(11;14)(q13;q32) popisovaná u leukémie plášťových buněk a myelomů. Při této fúzi je 5'konec CCND1 translokován na chromosom 14 do oblasti genu pro těžký řetězec imunoglobulinu - IgH. Přestavba má za následek změnu v promotoru genu, což vede k zvýšené expresi cyklinu D1. Translokace bývá vždy spojena se špatnou prognózou a sníženou dobou přežití nemocných (Levy a kol. 1999). ATM (ataxia telangiectasia mutated) Delece genu ATM byly prokázány u chronické lymfatické leukémie, jejímž rysem je vysoká invazivita a proliferace lymfatických buněk (Winrow a kol. 2005). Poprvé byly mutace ATM popsány u autosomálně recesivního onemocnění ataxia telangiectasia, charakterizovaném neuromuskulárními poruchami, imunodeficiencí, zvýšenou senzitivitou k radiačnímu záření, chromosomovou nestabilitou a vysokým rizikem vzniku lymfoidních malignit. Jejich studiem byly zjištěny mutace a delece genu ATM v obou alelách, proto je považován za nádorový supresorový gen (Stilgenbauer a kol. 1997). Gen ATM je lokalizován v oblasti 11q22.3. Obsahuje 66 exonů o celkové délce 184 kb. Gen je exprimován ve všech tkáních. Kóduje jaderný fosfoprotein o aminokyselinách a velikosti 350 kda. Tato serin/treoninová proteinkináza hraje 19
20 důležitou roli při reakci buňky na ionizující záření, které způsobuje dvouřetězcové zlomy DNA. Autofosforylací se inaktivní dimer rozdělí na aktivní monomery ATM, které fosforylují další proteiny. Mezi známé substráty kinázy patří proteiny p53, BRCA1 a mnoho dalších účastnících se reparace DNA a regulace buněčného cyklu. To vše má za následek aktivaci reparačních mechanizmů a snahu pozastavit cyklus při poškození DNA ionizačním zářením (Chen a kol. 2005). Vorechovsky a kol. (1997) prokázali jako místo nejčastější mutace vysoce konzervativní oblast kódující kinázovou doménu proteinu. Důsledkem mutace je změna vazebné oblasti pro adenosintrifosfát či substrát Změny krátkého ramene chromosomu 11 (11p) Abnormality krátkého ramene chromosomu 11 se u hematologických malignit vyskytují spíše jen vzácně. Obvykle postihují oblast 11p15 a zde lokalizovaný gen NUP98 (nucleoporin 98kDa). Další popsáné alterace krátkého ramene jsou uvedeny v tabulce 3. Tabulka 3. Alterované geny na 11p u hematologických malignit. Oblast Gen Funkce Aberace Onemocnění 11p15 LMO1 transkripční faktor t(11;14)(p15;q11) T-buněčné ALL 11p15.4 NUP98 jaderný transport přestavby 11p15 AML, MDS, ALL 11p13 LMO2 regulátor hematopoesy t(11;14)(p13;q11), t(7;11)(q35;p13), del(11)(p12p13) T-buněčné ALL Vysvětlivky: MDS (myelodysplastický syndrom), AML (akutní myeloidní leukémie), ALL (akutní lymfatická leukémie). NUP98 (nucleoporine 98 kda) První změna genu NUP98 byla identifikována v roce 1996 (Borrow a kol. 1996). Od té doby byly mutace NUP98 jako vzácné rekurentní abnormality popsány u AML, MDS, CML, T-buněčné ALL, a to primárních i sekundárních typů (Nebral a kol. 2005, Zhang a kol, 2007, Wang a kol. 2010). Dle obsáhlé mezinárodní studie zabývající se sekundárními leukémiemi jsou nejčastějšími změnami inverze (11)(p15q23) a translokace t(7;11)(p15;p15). Jako přídatné aberace se objevují delece dlouhých ramen chromosomů 7 a 5 nebo monosomie 7 (Rowley a Olney, 2002). Vzhledem k své velmi distální lokalizaci je řada přestaveb genu NUP98 klasickou cytogenetickou analýzou nedetekovatelná, pro identifikaci je proto třeba citlivějších molekulárních metod. 20
 a RNA vazebný motiv na C-konci.")
21 Gen kóduje protein o 920 aminokyselinách, velikosti 98 kda. Obsahuje repetitivní motiv na N-konci (FG, FxFG nebo GLFG; G-glycin, L-leucin, F-fenylalanin, x- jakákoliv aminokyselina) a RNA vazebný motiv na C-konci. Protein se nachází v jaderné membráně savců, kde v asociaci s dalšími nukleoporiny tvoří supramolekulární strukturu důležitou pro jaderný transport (Iwamoto a kol. 2010). Jeho role v maligní transformaci není zcela objasněna, neboť jeho vysoce specializovaná funkce nezahrnuje vývoj krevních buněk. Zdá se tak, že významnou úlohu v nádorovém zvratu mají partnerské fúzní geny, které vlivem translokace vytvářející spolu s N-koncem NUP98 chimérický protein. Hypotézu potvrzuje fakt, že většina těchto genů patří mezi geny homeoboxu (až 50 % případů) (Kozev a kol. 2004). Do roku 2010 bylo identifikováno 29 různých fúzních partnerů (viz obrázek 3). Z důvodu vzácného výskytu NUP98 přestaveb je jednoznačné stanovení prognózy zatím obtížné. Recentní studie poukazují na spojitost spíše se špatným klinickým průběhem (Hidaka a kol. 2007, La Starza a kol. 2009, Hollink a kol., 2011, Lundin a kol., 2011). Obrázek 3. NUP98 gen a jeho partnerské geny. Převzato z k datu
22 3.4. Akutní myeloidní leukémie (AML) Myeloidní leukémie patří mezi nádorová onemocnění krvetvorné tkáně, které postihují buňky myeloidní řady. Akutní formy této nemoci jsou charakterizovány akumulací nezralých krevních elementů v kostní dřeni. Abnormální buňky patologického klonu mají omezenou schopnost diferenciace a vykazují různé funkční poruchy. Zdravá krvetvorba bývá potlačena. Zpočátku se AML projevuje nespecificky. U nemocných se objevuje únava a krvácivé nebo infekční komplikace. Mezi symptomy patří horečka, ztráta hmotnosti, nechutenství, chudokrevnost nebo bolesti kostí (Adam a kol. 2008). Nemoc bývá ročně diagnostikována u cca 3: obyvatel a tato incidence se v posledních letech nemění. Převažuje u dospělých, kde tvoří 80 až 85 % všech akutních leukémií u osob starších 20 let. Častěji postihuje obyvatele větších průmyslových měst. Medián vzniku tohoto onemocnění se pohybuje mezi 63 až 65 lety (Mayer a Starý 2002). V dětské populaci se objevuje relativně vzácně. Představuje asi jen % dětských akutních leukémií. Jde především o děti velmi nízkého věku tj. mladších 1 rok (Hall 2001). Charakteristickým znakem AML je v krvi zjistitelná normocytární anémie, trombocytopenie a leukocytóza, zpravidla s vyplavením blastických elementů (> 20 % myeloblastů). Obvyklá bývá i infiltrace jednotlivých orgánů jako jsou játra, slezina, kůže nebo centrální nervový systém, což vede k poruše jejich funkce. Vzácně může dojít k vzniku granulocytárního sarkomu. Akutní myeloidní leukémii také relativně často provází trombo - hemoragické komplikace (Adam a kol. 2008). AML může vzniknout de novo nebo vlivem předešlé léčby, obzvláště antitopoizomerázními léky jako jsou etoposidy nebo teniposidy (Sung a kol. 2006). K vzniku leukémie také dochází po vystavení organismu některým chemickým látkám jako je benzen nebo vlivem přítomnosti některých virů. Leukémie tohoto typu se nazývají sekundární. Rizikovými faktory pro vznik akutní myeloidní leukémie je kromě ionizujícího záření a chemoterapeutické cytostatické léčby také předchozí myeloproliferativní onemocnění nebo myelodysplastický syndrom, který se vyskytuje poměrně vzácně především u starších lidí a v AML přechází u 25 až 30 % nemocných (Mayer a Starý 2002, Adam a kol. 2008). Zvýšené riziko vzniku AML mohou mít i některá vrozená genetická onemocnění jako je například Downův syndrom nebo syndromy chromosomové nestability. V současné době se k diagnostice AML využívá 22
23 cytomorfologie, konvenční cytogenetika, interfázní a metafázní fluorescenční in situ hybridizace (FISH), polymerázová řetězcová reakce (PCR) a imunofenotypizace. Klasifikovat AML je možno na základě 2 systémů: staršího FAB (French- American-British), který rozeznává 8 subtypů AML (M0 až M7) na základě morfologie a vyzrávání buněk (viz Příloha 1), a novějšího WHO (World Health Organization), který zohledňuje nejen morfologické hodnocení, ale i cytogenetické výsledky vyšetření kostní dřeně a dalších doplňujících vyšetření (Swerdlow a kol. 2008, viz Příloha 2). Snahou léčby leukémie je dosáhnout kompletní remise, což je stav, kdy pacient nepociťuje žádné příznaky nemoci a nemoc není diagnostikovatelná rutinními hematologickými metodami. To znamená, že nález leukemických buněk v kostní dřeni nepřesahuje 5 %. V 70. a 80. letech došlo k zavedení kombinované chemoterapie do léčby AML nemocných, založené na indukci daunorubicinem (DNR) a cytosin arabinosidem (Ara-C). Frekvence kompletní remise u nemocných se sice zvýšila, přesto však nedosahovala ani 50 %. Také k dlouhodobému přežití (více než 3 roky) docházelo velmi sporadicky. Jelikož v krvi zůstává stále malé množství leukemických buněk, nazývané minimální zbytková choroba (MRD), i při kompletní remisi, je vysoká pravděpodobnost, že bez další léčby nastane relaps. V 90. letech byla proto do léčby AML zařazena vysokodávkovaná konsolidace, která se přínosně projevila na dlouhodobém přežívání pacientů. Nevýhodou ale byla výrazná toxicita terapie. V 90. letech také došlo k zavedení alogenní transplantace krvetvorných buněk do léčebného programu AML nemocných. Od roku 2002 byla léčba volena na základě stratifikace nemocných do prognostických skupin dle cytogenetického nálezu. Indukce spočívala v kombinaci Idarubicinu (10 mg/m 2, den 1-3) a Ara-C (100 mg/m 2, den 1-7). K udržení navozené remise se podávala konsolidace (3 mg/m 2 Ara-C v den 1, 3 a 5). V případech vysokého rizika byla indikována transplantace kostní dřeně. Od roku 2010 je léčebný postup volen nejen na základě cytogenetického, ale i molekulárně-genetického nálezu a léčba je dále mofikována dle monitorace molekulárně genetických markerů minimální zbytkové choroby. Standardně je k indukci remise u AML pacientů mladších 60 let používán režim s DNR 90 mg/m 2 podávaným tři dny v kombinaci s Ara-C 100 mg/m 2 podávaným sedm dní. Starší pacienti jsou léčeni modifikací s Idarubicinem 10 mg/m 2. Frekvence dosažení kompletní remise u AML pacientů tak vzrostla na 70 % (Soukup a kol. 2012). Většině nemocných se středním a vysokým rizikem se však leukémie vrací do 3 let od stanovení diagnózy (Döhner a kol. 2010). Prognóza po relapsu bývá velmi nepříznivá a jediným kurativním přístupem zůstává transplantace kostní dřeně. 23
24 Chromosomové změny u AML a jejich prognostický význam Akutní myeloidní leukémie je nejen fenotypicky, ale i geneticky velmi heterogenní onemocnění. Mitelman ve své databázi uvádí více než 200 různých strukturních a numerických chromosomových aberací popsaných u AML ( k datu ). Abnormální karyotyp lze sledovat u 50 až 60 % nemocných s AML (Grimwade a kol. 2001, Mrózek a kol. 2004, Marchesi a kol. 2011). Komplexní změny karyotypu (tři a více chromosomových abnormalit) se vyskytují u 10 až 15 % případů (Mrázek a kol. 2008, Marchesi a kol. 2011). Nejčastěji bývají jejich součástí chromosomy 5, 7, 8, 10, 11, 12, 17 a 21 (Babická a kol. 2007). Hyperdiploidie u AML nebývá příliš častá. Obvykle zahrnuje chromosomy 4, 8, 10, 11, 13, 14, 19 a 21, přičemž trisomie chromosomu 8 následovaná trisomií chromosomu 21 se vyskytují nejvíce (Luquet a kol. 2007). Různé chromosomové aberace jsou popisovány i u jednotlivých morfologických FAB subtypů. Tyto přestavby mají diagnostický a prognostický význam a lze je využít k monitorování průběhu onemocnění a k detekci minimální zbytkové choroby (viz tabulka 4) (Haferlach a kol. 2003, Marcucci a kol. 2004, Grimwade a kol. 2010). Tabulka 4. Výskyt chromosomových změn u jednotlivých FAB subtypů AML. Chromosomová aberace FAB Výskyt Prognóza subtyp v subtypu t(8;21)(q22;q22) M2 13 % příznivá t(15;17)(q22;q21) M3 95 % příznivá inv(16)(p13q22) M4 10 % příznivá aberace 11q23 M4,M % nepříznivá t(9;11)(p22;q23) M5 20 % střední inv(3)(q21q26) M2, M4, M7 3 5 % nepříznivá Mezi nejčastější chromosomové aberace bez ohledu na AML FAB subtyp patří abnormality chromosomů 5 a 7 (delece dlouhých ramen nebo monosomie), 11 a trisomie chromosomu 8 (Marchesi a kol. 2011). Typickým nálezem jsou především pro sekundární formu AML. Zatímco abnormality chromosomu 11, zvláště oblasti q23 a zde lokalizovaného genu MLL, jsou spojovány s inhibitory topoizomerázy II, u nemocných léčených pomocí alkylačních látek nebo po expozici benzenu se objevují delece dlouhých ramen chromosomů 5 a 7 nebo monosomie chromosomu 7 (Escobar a kol. 2007, Tennant a kol. 2007, Qian a kol. 2010). V současné době slouží řada cytogenetických abnormalit nejen jako diagnostické markery pro stanovení specifického typu AML, ale i jako nezávislé prognostické 24
25 faktory využívané ke sledování dosažení kompletní remise, rizika relapsu a celkového přežití (Mrózek a kol. 2007, Marchesi a kol. 2011). Dle cytogenetického nálezu lze AML rozdělit do 3 odlišných prognostických skupin (viz tabulka 5, obrázek 4). První skupinu tvoří pacienti s balancovanými přestavbami: translokací t(15;17)(q22;q21), t(8;21)(q22;q22) a t(16;16)(p13;q22) nebo pericentrickou inverzí inv(16)(p13q22). Jsou asociovány s dobrou odpovědí na chemoterapii a dlouhou dobou přežití. Díky svému specifickému charakteru, nespadají tyto abnormality dle WHO klasifikace do skupiny komplexního karyotypu ani tehdy, vyskytují-li se s dalšími, přídatnými chromosomovými změnami (Döhner a kol. 2010, Marchesi a kol. 2011). Translokace t(15;17)(q22;q21) je ukázkou významu objasnění molekulární podstaty chromosomových změn. Identifikací alterovaného genu, jímž je receptor kyseliny retinové RARA (17q21), došlo k zavedení cílené terapie pro nemocné s touto translokací, a to pomocí inhibitorů kyseliny retinové tvz. ATRA léčba. Z dřívě prognosticky nepříznivého onemocnění se tak v současnosti stalo prognosticky velmi příznivé, dobře léčitelné (Adam a kol. 2008). Druhou skupinu představují pacienti s normálním karyotypem, translokací t(9;11)(p22;q23), trisomií chromosomu 8 a dalšími chromosomovými změnani, které nejsou řazeny do první nebo třetí prognostické skupiny. Klinický stav těchto nemocných je lepší než nemocných z třetí skupiny, ale horší než ze skupiny první (Grimwade a kol. 2010, Marchesi a kol. 2011). Třetí skupina je charakteristická velmi nepříznivou prognózou spojenou s celkově krátkou dobou přežití a špatnou odpovědí na léčbu, pokud se podaří dosáhnout remise je velmi vysoká pravděpodobnost relapsu (Grimwade a kol. 2010, Grimwade a Mrózek 2011, Marchesi a kol., 2011). Jsou do ní řazeny komplexní změny karyotypu, t(3;3)(q21;q26), inv(3)(q21;q26), t(6;9)(p23;q34), -7, del(5q) a přestavby genu MLL kromě t(9;11)(p22;q23). Grimwade a kol. (1998) ještě řadil všechny abnormality MLL genu ke středně příznivé prognóze, ale mnoho pozdějších studií, zabývající se jednotlivými MLL translokacemi, poukázaly na jejich spojitost s prognózou velmi nepříznivou (Schoch a kol. 2003, Grimwade a kol. 2010, Marchesi a kol. 2011). Výjimku tvoří pouze translokace t(9;11)(p22;q23), která zůstala v druhé prognostické skupině. Podobně jako je tomu u prognosticky příznivých balancovaných změn ani prognosticky nepříznivé balancované abnormality nespadají dle nové WHO klasifikace do skupiny komplexního karyotypu, a to i přes přítomnost dalších aberací (Döhner a kol. 2010, Marchesi a kol. 2011). 25

26 Pro pacienty s dobrou prognózou představuje léčba obvykle několik dávek intenzivní chemoterapie. U pacientů s nepříznivou prognózou se doporučuje transplantace kostní dřeně, stejně jako u pacientů s relapsem AML. U nemocných se středně příznivou prognózou se rozhoduje na základě konkrétní situace (Dohner a kol. 2010). Tabulka 5. Prognostický význam aberací u AML (Marchesi a kol. 2011). Obrázek 4. Křivky přežití pro vybrané abnormality (Grimwade a Hills 2009). 26
. Snahou řady studií je proto najít vhodné molekulární prognostické markery i u této skupiny nemocných.")
27 Molekulární markery u AML a jejich prognostický význam Literatura udává 40 až 50 % případů AML s normálním karyotypem určeným klasickou cytogenetickou analýzou (Grimwade a kol. 2001, Mrózek a kol. 2004, Marchesi a kol. 2011). Snahou řady studií je proto najít vhodné molekulární prognostické markery i u této skupiny nemocných. Jejich přehled a prognostický význam je zobrazen na obrázku 5. Nejčastější genetickou abnormalitou u AML s normálním karyotypem (NK) jsou mutace genu pro jaderný fosfoprotein nukleofosmin 1 (NPM1; 5q35). Nemocní s mutací tohoto genu mají lepší podíl úplné remise a celkového přežití. Je tomu tak ale jen v případě, že se nevyskytuje současně s mutací tyrozinkinázy 3 (FLT3; 13q12), která je druhou nejčastější genetickou změnou u AML s NK. Její interní tandémové duplikace (ITD) jsou spojovány s velmi nepříznivou prognózou a zhoršují klinický stav i u nemocných, u nichž byly současně prokázány jinak prognosticky příznivé abnormality. Doba trvání remise a celkové přežití jsou delší u nemocných s mutací genu pro transkripční faktor CEBPA (19q13), zvláště jde-li o mutaci bialelickou. Dalším důležitým alterovaným genem u AML s NK je i již zmiňovaný gen MLL (11q23) a jeho parciální tandémové duplikace. MLL PTD byly prvním molekulárním markerem u AML s NK spojeným se špatnou prognózou pro nemocné. Ve 30 až 40 % případů se vyskytují spolu FLT3-ITD. Mezi další studované geny na molekulární úrovni u AML patří TP53, WT1, RAS, IDH1, IDH2, DNMT3A, TET2, BAALC, ERG, MN1, EVI1, BCOR, atd (Ghanem a kol. 2012, Grossmann a kol. 2012, Haferlach a kol. 2012). V současné době se výzkum zaměřuje především na mutační skríning vybraného panelu genů pomocí metod sekvenace. Obrázek 5. Frekvence a prognostický význam vybraných molekulárních markerů u AML (převzato od Grimwade a Hills, 2009). 27
28 AML u dětí Dětská AML tvoří jen % ze všech dětských leukémií. Oproti dospělým nemocným s AML je u dětských pacientů patologický karyotyp prokázován až v 80 % případů (Mrózek a kol. 2004, Manola 2009). Dle věku, AML FAB subtypu a cytogenetického nálezu lze dětskou AML rozdělit do dvou kategorií. První skupinu tvoří mladší děti, u kterých převažují FAB skupiny M4, M5 a M7. Cytogenetický nález bývá oproti starším dětem a dospělým více specifický. Mezi nejčastější chromosomové změny patří přestavby MLL genu (11q23.3), jejichž incidence roste s klesajícím věkem (nejvyšší u dětí do 3 let) (Manola 2009). Dle obsáhlé studie MLL přestaveb u dětí byla prokázána příznivější prognóza pro translokaci t(1;11)(q21;q23), zatímco t(6;11)(q27;q23), t(10;11)(p12;q23) a t(10;11)(p11.2;q23) byly identifikovány jako nezávislý ukazatel nepříznivého klinického průběhu. Navzdory řadě analýz prokazujících lepší prognózu translokace t(9;11)(p22;q23) u AML se v této studii autorům nepodařilo spojitost potvrdit (Balgobind a kol. 2009). U dětí mladších 1 roku byly sledovány reciproké translokace, které dosud nebyly popsány u dospělých a jsou tak výlučně spjaty s nízkým věkem pacienta. Jedná se o translokace t(1;22)(p13;q13) a t(7;12)(q36;p13), která bývá zaměnována za del(12)(p13). U obou změn byl prokázán nepříznivý klinický průběh. Významnou část mladších dětí s AML tvoří děti s Downovým syndromem, tedy vrozenou trisomií chromosomu 21. Z morfologického hlediska odpovídá jejich AML vždy typu akutní megakaryocytární leukémie AML-M7. U 10 % dětí s Downovým syndromem se již velmi časně objevuje transientní myeloproliferace s mutací genu GATA1 (Xp11.23). V 80 % případů dojde k spontánnímu vymizení nemoci, ve zbylých 20 % dochází vlivem dalších mutací k přechodu do AML-M7. Na chromosomu 21 byla identifikována řada důležitých genů uplatňujících se v hematopoese (např. RUNX1, ERG, ETS2, mir99a, mir125b), přídatně byly sledovány mutace genů JAK3, JAK2, TP53, FLT3 a dalších (Khan a kol. 2011). Starší dětí, u kterých často převažuje FAB subtyp M0, M1, M2 a M3, tvoří druhou skupinu dětské AML. Výsledky cytogenetické analýzy se více shodují s nálezy dospělých nemocných. Mezi časté chromosomové změny patří prognosticky příznivé abnormality t(8;21)(q22;q22), t(15;17)(q22;q21) a inv(16)(p13q22) (Mrózek a kol. 2004). Z početních odchylek bývají detekovány trisomie chromosomů 8, 21 a 22. Delece del(5q), del(7q)/-7, abnormality chromosomu 3 nebo komplexní změny karyotypu se oproti dospělým vyskytují u dětí spíše vzácně. Spojeny jsou stejně jako u 28
29 starších AML pacientů s nepříznivou prognózou, kromě delece del(7q), u které se prokázal lepší klinický průběh než u úplné monosomie 7. Důvodem by mohl být vyšší výskyt del(7q) spolu s prognosticky příznivými aberacemi. Je tak doporučeno tyto dvě abnormality u dětí odlišovat (Manola 2009). Mezi kryptické změny, často nedetekovatelné klasickou cytogenetickou analýzou, patří přestavby genu NUP98 (11p15). Translokace t(5;11)(q35;p15) při zdánlivě normálním karyotypu nebo del(5q) byla poprvé popsána právě u dětí (Jaju a kol. 2001). Teprve později ji další analýzy potvrdily i u dospělých nemocných (Casas a kol. 2003). Vzhledem k vzácnému výskytu NUP98 alterací, nebyl konečný prognostický význam řady těchto translokací zatím stanoven. V současné době se klinický průběh onemocnění u dětí i dospělých s touto abnormalitou jeví spíše jako nepříznivý. Za posledních 20 let se 5leté přežití dětských pacientů s AML zvýšilo z původních 30 % na 65 %. I navzdory intenzivní terapii relabuje a zemře až polovina nemocných dětí s AML. Značným současným problémem je také stále poměrně vysoká úmrtnost na vysokodávkovou léčbu (Manola 2009). Cílem recentních výzkumů je proto vyvinutí cílené terapie s minimálním poškozením zdravých tkání. 29

30 4. MATERIÁL A METODY 4.1. Materiál Soubor pacientů Vyšetřovaný soubor byl sestaven z nemocných s nově diagnostikovanou AML, kteří byli od ledna 2006 do května 2012 vyšetřeni v cytogenetické laboratoři ÚHKT v Praze. Od ledna 2009 do května 2012 byli do souboru zahrnuti i pacienti, jejichž vzorek kostní dřeně/periférní krve byl zaslán k cytogenetické analýze do Centra nádorové cytogenetiky ÚLBLD VFN a 1. LF UK. Všichni nemocní poskytli informovaný souhlas pro využití materiálu pro vědecké účely. Diagnóza AML byla stanovena dle FAB a/nebo WHO (2008) nomenklatury. Ze studie byli vyloučeni nemocní s akutní promyelocytární leukémií (dle FAB AML-M3), která tvoří velmi specifický typ AML s odlišnou léčbou. Celkem jsme vyšetřili buňky kostní dřeně/periferní krve u 300 dospělých nemocných. Jednalo se o 159 mužů a 141 žen (průměrný věk 55 let, rozpětí 19 až 84 let). Primární typ AML byl diagnostikován u 229 nemocných, v 71 případech šlo o sekundárně vzniklou leukémii, převážně transformovanou z myelodysplastického syndromu, vzácněji na podkladě lymfomu nebo pravé polycytemie. Z karcinomů se nejčastěji jednalo o rakovinu prsu a ovarií u žen a rakovinu testes u mužů. Celkové zastoupení jednotlivých AML FAB subtypů je uvedeno v grafu 1. Dle WHO odpovídala AML převážně typu s multilineární dysplázií a blíže nespecifikované. Během sledovaného období bylo v Centru nádorové cytogenetiky vyšetřeno i 14 dětí s nově diagnostikovanou primární AML. Dětský soubor zahrnoval 7 dívek a 7 hochů (průměrný věk 6,4 let, rozpětí od 1 roku do 15 let). Dle FAB subtypu se jednalo o AML: M5 (5x), M4 (4x), M2 (2x), M0 (2x) a M7 (1x, ve spojitosti s Downovým syndromem). Graf 1. Zastupení jednotlivých AML FAB subtypů u dospělých nemocných. 30
31 4.2. Metodika Klasická cytogenetická analýza Všichni nemocní (dospělí i děti) byli vyšetřeni klasickou cytogenetickou analýzou, při níž byl sestaven karyotyp dělících se maligních buněk (kostní dřeně nebo periférní krve) podle závazné mezinárodní nomenklatury ISCN (Shaffer a kol. 2009). Buňky získané při odběru byly zpracovány standardními technikami (kultivace v MarrowGrow (buňky kostní dřeně) nebo LymphoGrow (buňky periférní krve) médiu, přidání kolcemidu, hypotonie, fixace a barvení Wright odpovídající G-pruhům). Pomocí IKAROS zobrazovacího systemu pro karyotypování (MetaSystems TM ) bylo zhodnoceno 22 mitos, pokud byly na preparátech přítomny Molekulární cytogenetická analýza Metoda FISH Vzhledem ke kryptickému charakteru některých přestaveb MLL genu (11q23.3), byli dospělí i děti vyšetřeni metodou FISH s dvoubarevnou lokus-specifickou sondou pro tento gen LSI MLL Break Apart Rearrangement probe (Abbott Vysis TM ) (viz obrázek 6 a 7). U 245 dospělých pacientů bylo FISH vyšetření rozšířeno o sondy detekující další nejčastější chromosomové změny u AML: deleci dlouhých ramen chromosomu 7, oblasti 7q31/monosomii chromosomu 7 (sonda LSI 7q31/CEP 7), deleci dlouhých ramen chromosomu 5, oblasti 5q31 (sonda LSI 5q31/5p15.2) a početní odchylky chromosomu 8 (sonda CEP 8/kontrola obvykle centromerická sonda pro chromosom 9 (CEP 9)). K potvrzení nebo upřesnění patologického nálezu klasické analýzy byly použity další lokus-specifické, centromerické a/nebo subtelomerické sondy (Abbott Vysis TM, Kreatech TM, MetaSystems TM ). Obrázek 6. Schéma LSI MLL sondy (Abbott Vysis TM ). BCR (breakpoint cluster region). Převzato z stav k
32 Zlomová místa na chromosomu 11 byla analyzována FISH metodou s komerčně dodávánými sondami LSI ATM (11q22) a LSI CCND1 (11q13) (Abbott Vysis TM ) a především s vybranými BAC (bacterial artificial chromosome) (BlueGnome) sondami (v kombinaci s centromerickou sondou pro chromosom 11 (CEP 11) nebo subtelomerickými sondami (ToTel 11p nebo ToTel 11q) (Abbott Vysis TM ). Přehled použitých BAC sond je uveden v tabulce 5. Při FISH analýze jsme postupovali dle doporučení výrobce. Z důvodu možné zkřížené hybridizace (cross-hybridization) BAC sond a pro kontrolu jejich správné lokalizace na chromosomu, jsme všechny BAC sondy nejprve otestovali hybridizací k normálním chromosomům buněk periferní krve. V případě zkřížené hybridizace byly odečteny pouze mitosy. Tabulka 5. Přehled použitých BAC sond pro mapování chromosomu 11. (pozice bází dle UCSC, hg19) Oblast 11p p p13 11p12 11p11.2 BAC sonda (BlueGnome) RP11-348A20 RP11-92N11 RP11-258P13 RP11-120E20 Lokalizace RP11-406D RP11-244O RP11-242P RP11-258H RP4-562D RP11-140L RP11-366N RP-11-26B RP11-74J RP11-98C RP11-68D RP11-514F RP11-465C RP11-233I RP11-144G RP11-160M RP11-40H RP11-454H RP11-375D RP11-538D RP11-108L RP11-484D RP11-70A RP11-450F RP11-12C RP11-164L RP11-433B RP11-425L RP11-368A RP11-165B RP11-169J RP11-390K RP11-17G RP11-125F Oblast BAC sonda (BlueGnome) Lokalizace 11q13.1 RP11-472D RP11-424B4 RP11-715F q13.2 RP11-98N RP11-105D RP11-569N RP11-554A q13.4 RP11-92H q14.1 RP11-139M8 RP11-15N10 RP11-7H RP11-16H RP11-153F q14.2 RP11-241M RP11-496E q14.3 RP11-135L RP11-54L q21 RP11-159N RP11-277H q22.1 RP11-16K RP11-379J RP11-179B RP11-22J q22.3 RP11-273B RP11-268L RP11-18B q23.1 RP11-285P q23.2 RP11-267J q23.3 RP11-215H Oblast BAC sonda (BlueGnome) Lokalizace 5q35 RP11-265K p21.31 RP11-175A q12 RP1-1J RP3-387E
33 K vyhodnocení zhotovených preparátů jsme používali fluorescenční mikroskop AXIOPLAN 2 Imaging (Zeiss), vybavený specifickými optickými filtry pro detekci použitých fluorochromů. Na každém preparátu jsme počítali fluorescenční signály alespoň ve 20 mitosách (pokud jich bylo tolik přítomno pod krycím sklem) a ve 200 interfázních jádrech. Vyhodnocovali jsme počet a charakter fluorescenčních signálů, v mitosách navíc jejich lokalizaci na chromosomu. Cut-off level byla stanovena pro translokace, duplikace a amplifikace na 2,5 % a pro delece a monosomie na 5 %. Nález jsme popsali podle mezinárodní ISCN nomenklatury (Shaffer a kol. 2009). Patologický klon jsme z fluorescenčního mikroskopu zdokumentovali CCD kamerou a zpracovali pomocí softwaru ISIS (MetaSystemsTM). Metody mfish, mband Metoda mfish byla použita k analýze mnohočetných změn chromosomů. Používali jsme celochromosomové sondy značené fluorochromy: FITC, Spectrum Orange TM, TexasRed, DEAC a Cy TM 5. Metodu mband jsme zvolili k zpřesnění lokalizace zlomových míst a k detekci oblastí amplifikace/delece. V obou případech jsme používali DNA sondy a detekční kity od firmy MetaSystems TM, postupovali jsme dle doporučení výrobce. Metody mikroarray Nebalancované změny genomu byly analyzovány metodami mikroarray. V 10 případech jsme použili SNP array (HumanCytoSNP-12 BeadChips, Illumina). DNA byla z fixovaných buněk kostní dřeně/periferní krve izolována užitím QIAamp DNA mini Kitu (Qiagen). Všechny metody byly provedeny dle instrukcí výrobce Molekulární genetická analýza U 210 dospělých nemocných ze souboru byla provedena také molekulárně genetická analýza pro zjištění přítomnosti parciálních tandemových duplikací genu MLL. Izolace RNA a reverzní transkripční (RT) PCR RNA byla z buněk kostní dřeně a/nebo periferní krve, vyizolována pomocí Ficoll- Paque PLUS (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) dle doporučení výrobce. K syntéze komplementární DNA byla použita SuperScript II reverzní 33
34 transkriptáza (Invitrogen Corporation, Carlsbad, CA, USA). První kolo PCR proběhlo s primery MLLex7-F 5 -GGAAGTCAAGCAAGCAGGTC-3 a MLLex3-R 5 - AGGAGAGAGTTTACCTGCTC-3, druhé s primery MLLex8-F 5 - GTCCAGAGCAGAGCAAACAG-3 a MLLex3-RII 5 - ACACAGATGGATCTGAGAGG-3 (viz obrázek 7). PCR produkty byly následně zkontrolovány elektrofórezou (2% agarosový gel). DNA sekvenace PCR produkty získané z gelu elektroelucí byly přečištěny a osekvenovány v obou směrech (BigDye Terminator v1.1 Cycle Sequencing Kit; Applied Biosystems) na analyzátoru ABI (Applied Biosystems). Obdržené sekvence byly vyhodnoceny dle parametrů genové banky NM Obrázek 7. Exon-intronová struktura genu MLL. Vysvětlivky: umístění sond pro FISH (5 konec MLL - zelená šipka, 3 konec MLL - oranžová šipka) a primerů užitých pro 1. a 2. kolo PCR. Porovnání klasifikací exonů MLL genu: NM_ (námi užitá) a dvě starší verze [A] dle Strout a kol. 1998; [B] dle Nilson a kol Převzato od Sarova a kol. (2009) Statistická analýza Statistické vyhodnocení bylo provedeno pomocí Kaplan-Mayerovy analýzy přežívání s Mantel-Coxovým testem přežívání. Jako kontrolní byl použit soubor nemocných s normálním karyotypem (>10 normálních mitos, negativní FISH skríningový panel a negativní MLL PTD). U žijících nemocných byla doba přežití stanovena k datu
35 5. VÝSLEDKY U všech 300 dospělých a 14 dětských nemocných s nově diagnostikovanou AML byla provedena klasická cytogenetická analýza. U dvou dospělých nemocných jsme prokázali netypickou reciprokou translokaci přítomnou ve všech sledovaných mitosách. Předpokládali jsme proto, že se jedná o vrozené chromosomové abnormality. U jednoho nemocného jsme hypotézu ověřili detekcí této změny v dělících se buňkách periferní krve kultivovaných 72h s přídavkem phytohemagglutininu (PHA). U druhého jsme kontrolní materiál k ověření neobdrželi. Na základě klasické a molekulárně cytogenetické analýzy jsme patologický nález prokázali u 211 dospělých nemocných (70 %) (viz graf 2). U 38 (13 %) pacientů nebyly přítomny dělící se buňky (nález 0 mitos). Mezi nejčastější strukturní abnormality patřily: přestavba MLL genu (n=28), del(7)(q31)/-7 (n=16), t(8;21)(q22;q22) (n=9), inv(16)(p13q22)/t(16;16)(p13;q22) (n=8), del(5)(q31) (n=5) a t(3;3)(q21;q26)/ins(3;3)(q26;q21q26) (n=3). Nejčastějšími početními změnami byly +8, +13, +21 a +11. Komplexní karyotyp jsme prokázali u 51 nemocných (17 %). Jeho součástí byly obvykle chromosomy: 5, 17, 7, 21, 12, 11, 20, 8, 10 a 3. Graf 2. Cytogenetický nález u dospělých nemocných s nově diagnostikovanou AML. 102 (34%) 51 (17%) 109 (36%) 38 (13%) Normální nález 0 mitos Komplexní karyotyp Ostatní Kombinací klasické a molekulárně cytogenetické analýzy jsme získané chromosomové aberace prokázali u 12 dětských nemocných (86 %). U 2 (14 %) pacientů nebyly přítomny dělící se buňky (nález 0 mitos). Nejčastější strukturní abnormalitou byla přestavba MLL genu (n=5), sporadicky se vyskytly: del(7)(q31) (n=1), t(8;21)(q22;q22) (n=1), inv(16)(p13q22) (n=1) a +13 (n=1). Komplexní karyotyp jsme prokázali pouze u jednoho nemocného. 35
36 5.1. Změny chromosomu 11 u dospělých nemocných s AML Změnu chromosomu 11 jsme prokázali u 55 (18 %) dospělých nemocných s nově diagnostikovanou AML. U jednoho nemocného se jednalo o reciprokou translokaci t(11;22)(q23;q11), přítomnou ve všech vyšetřovaných mitosách. Jelikož jsme translokaci potvrdili v PHA stimulovaných lymfocytech, uzavřeli jsme ji jako konstituční přestavbu chromosomů a pacient byl vyřazena z další analýzy. U pěti nemocných jsme prokázali trisomi chromosomu 11 bez přítomnosti zlomu (viz tabulka 6, nemocní č. 1 až 5). U jedenácti pacientů se na jednom chromosomu 11 nacházelo více zlomových míst; dva zlomy u čtyř nemocných (č. 11, 41, 44, a 54), tři a více zlomů u sedmi (č. 7, 8, 10, 42, 48, 52, 53). Ve dvou případech byly postiženy oba homologní chromosomy 11 (č. 10 a 42). Změna krátkého ramene chromosomu 11 (11p) byla prokázána u třinácti pacientů (č a 52-54). Alteraci dlouhého ramene chromosomu 11 (11q) jsme identifikovali u 40 pacientů (č ), u čtyř současně s abnormalitami krátkého ramene (č. 42, 52, 53, 54). Souhrn klinických dat nemocných a výsledků molekulárně cytogenetického mapování je shrnut v tabulce Mapování zlomů Na základě konvenční cytogenetiky a metod mfish/mband jsme rekurentní zlomová místa specifikovali do 12 různých chromosomových pruhů: 11p15 (n=7), 11p14 (n=2), 11p13 (n=6), 11p12 (n=1), 11p11.2 (n=6), 11p11.1 (n=2), 11q12 (n=2), 11q13 (n=3), 11q14-q21 (n=3), 11q22 (n=3), 11q23.3 (n=30) a 11q24 (n=3). Všichni nemocní byli vyšetřeni metodou FISH se sondou pro MLL gen (11q23.3). Vybrané oblasti jsme dále mapovali pomocí metody FISH se sondami pro geny ATM (11q22) a CCND1 (11q13) a serií BAC sond. U 10 nemocných byla také provedena SNP mikroarray. Z důvodu nedostatku materiálu nebyla dále mapována zlomová místa u nemocných č. 14 a 53. Výsledky a použití BAC sond zvolených pro mapování jednotlivých nemocných jsou uvedeny v tabulce 7. Užitím SNP array techniky byly nově odhaleny kryptické delece u čtyř nemocných č. 10, a 52. U čtyř nemocných (č. 13, 48, 50 a 52) bylo zlomové místo přehodnoceno do jiného pruhu, u jednoho nemocného (č. 45) zpřesněno ve stávajícím pruhu. U dvou nemocných (č. 40 a 41) s reciprokými přestavbami metoda neprokázala 36
37 žádné nebalancované změny chromosomu 11. Porovnání výsledků metod mband/mfish, mikroarray a FISH jsou uvedeny v tabulce 6. Metodou FISH jsme identifikovali další změny na chromosomu 11 u sedmi nemocných (č. 8, 10, 16, 18, 22, 26, 28). Lokalizace zlomových míst byla upravena užitím FISH mapování u devíti nemocných (č. 11, 12, 13, 44, 46, 47, 49, 50, 54), ve většině případů do sousedních chromosomových pruhů. U nemocného č. 47 nebyla kryptická delece, zjištěná metodou SNP mikroarray, potvrzena a zlomová místa tak nebyla započítána. U ostatních byl zlom upřesněn ve stávajícím pruhu identifikovaným metodou mband nebo mikroarry (viz tabulka 6). Pomocí FISH mapování jsme na chromosomu 11 identifikovali více než 35 různých zlomových míst, z toho dvě rekurentní (11p15.4 v genu NUP98 a 11q23.3 v genu MLL) a tři potenciálně nenáhodné v pruzích 11p13 (ch11: Mb), 11p12 (ch11: Mb) a 11q13.2 ( Mb) (pozice bází dle UCSC, hg19). Další překrývající se zlomová místa jsme detekovali i v oblastech 11p15.5 (ch11: Mb), 11p15.4-p15.1 (ch11: Mb), 11p12 ( Mb), 11q12 (nemapováno), 11q13.2 ( Mb), 11q14.2-q21 ( Mb) a 11q24 (nemapováno). Vzhledem k tomu, že tyto regiony nebyly z důvodu nedostatku materiálu dále upřesněny, zůstala oblast výskytu zlomu příliš veliká na to, abychom je mohly označit za jednoznačně rekurentní. V pěti případech pokrývavalo místo zlomu pouze jeden protein-kódující gen: NUP98 (11p15.4), MLL (11q23.3), LRRC4 (11p12), ODZ4 (11q14.1) a MAML2 (11q21). Přehled všech identifikovaných zlomových míst je zobrazen v tabulce tabulce 8. Hlavním mechanizmem vzniku zlomových míst na chromosomu 11 byly balancované přestavby, které jsme prokázali u 35 nemocných a jsou popsány v tabulce 9. Ve většině případů se jednalo o reciproké translokace genu MLL. Ostatní zlomy vznikly vlivem nebalancovaných změn. Oblasti duplikace a amplifikace Zmnožení genetického materiálu chromosomu 11 jsme pozorovali převážně na jeho dlouhém rameni. Parciální trisomie chromosomu 11 se vyskytla u osmi nemocných (č. 9, 14, 27, 44, 45, 46, 48, 50), přičemž u nemocné č. 44 jsme prokázali dva klony s duplikací chromosomu 11 o různé velikosti (viz tabulka 9 a obrázek 8 pod označením 44a a 44b). Společnou oblastí duplikace u všech nemocných byl region 11q q24 (n=9; ch11: Mb), začínající od 3 konce MLL genu. 37
38 Dalšími častými oblastmi duplikace byly 11q q23.3 (n=8; ch11: Mb), 11q21 11q22.3 (n=7; ch11: Mb) a 11q24 11qter (n=7; ch11: Mb). U žádného pacienta se nevyskytla parciální trisomie zahrnující pouze krátké rameno chromosomu 11. Amplifikaci oblastí chromosomu 11, tedy čtyři a více kopií, jsme prokázali u devíti nemocných (č. 7, 13, 18, 42, 48, 49, 51, 52 a 53). U nemocné č. 48 jsme v jednom klonu prokázali jak duplikaci (48a), tak i amplifikaci (48b) chromosomu 11 o různé velikosti. U nemocné č. 7 byla zmnožena pouze oblast na krátkém rameni chromosomu 11. V ostatních případech byly amplifikovanány současně i oblasti z dlouhého ramene. Minimálním úsekem amplifikace na 11q u všech nemocných byl 5 konec MLL genu (n=8). Dále byly často postiženy oblasti 11q q25 (n=7; ch11: Mb), 11q23.3 (n=7; ch11: Mb) a 11q q23.3 (n=6; ch11: Mb). Na krátkém rameni byl součástí amplikonu obvykle region 11p p15.1 (n=3). Přehled všech oblastí duplikace a amplifikace, jejich frekvence a kandidátní geny, které zahrnují, jsou uvedeny v tabulce 9 a obrázku 8. Oblasti delece Ztrátu genetického materiálu chromosomu 11 jsme popsali u devíti nemocných (č. 7, 8, 10, 11, 12, 42, 44, 48 a 52). Pouze u tří (č. 42, 44 a 48) byla deletována oblast na dlouhém rameni, v ostatních případech došlo k ztrátě části krátkého ramene chromosomu 11. V rámci 11p jsme neprokázali žádný společný deletovaný region. Některé se přesto vyskytovali častěji, a to 11pter-11p15.5 (n=4; ch11: Mb), 11p p13 (n=4; ch11: Mb) a 11p13 (n=4; ch11: Mb) (viz tabulka 9 a obrázek 8). 38
39 Tabulka 6. Klinická data nemocných s abnormalitou chromosomu 11 a výsledky molekulárně cytogenetického mapování. Pacient č. Pohl/ Datum Typ Zlomové místo na chromosomu 11 + nově detekované změny alo- Přežití Souhrný karyotyp (Klasická cytogenetická analýza a/nebo mfish/mband) Věk diagnózy AML mband SNP array (Mb) FISH (Mb) TKD (dny) TRISOMIE 11 1 Ž/ M1 47,XX,+11[22] - - nuc ish(mllx3)[36/200] ANO žije 53~55,XX,+X,+inv(X)(p21q12),+1+2,+4,del(5)(q13.3q33.1),t(8;12)(q24;q24),+10,+11, nuc ish(mllx3)[100/200] 2 Ž/ sm6 - - NE 193 der(12)t(12;13)(p11;q11),-13,+14,+21,+22[cp22] 42~45,XY,-3,der(4)t(4;17)(p31;?),der(5)t(5;12)(q12;p?),-7,t(8;9)(q?;p22),+11, nuc ish(mllx3)[24/200] 3 M/ M2 - - NE 22 der(12)t(12;17)(p?;?),der(12)del(12)(p11.2)del(12)(q21),-17,-18,+21[cp22] 4 M/ M0 47,XY,+11[3]/46,XY[14] - - nuc ish(mllx3)[18/200] NE 43 5 M/ saml 47,XY,+11[13]/46,XY[9] - - nuc ish(mllx3)[114/300] NE žije ABNORMALITY KRÁTKÉHO RAMENE CHROMOSOMU 11 (11p) 6 Ž/ sm6 43~48,XX,t(2;12)(p13;p13),del(5)(q13.3q33.3),t(5;11)(p13.3;p15),der(7)t(7;17) (q11;q12),-13,der(13)t(13;16)(p11;?),-17,-20,der(21)t(10;21)(q?;q11)[cp20]/46,xx[3] 11p15-11p15.5; ch11: NE žije 43~44,XX,del(5)(q13.3q33.3),der(11)del(11)(p11.2p14)inv(11)(p11.2p15.5)t(11;21) 7 Ž/ M4 (p15;q22),del(12)(p12p13),-13,der(13)t(13;21)(p11.1;q11.2)t(14;21)(q23;q22), 11p15 11p15.5; ch11: der(13)t(13;21)(p11.1;q11.2)t(17;21)(?;q22),der(14;14)(q10;q10),der(16)ins(16;11) 11p14 11p15; ch11: (?;p11.2p14)t(1;16;17)(?;p11.1;?),der(16)t(13;16)(q?;q12.1),-17,der(17)t(8;17) - 11p p11.2; ch11: (q13;p11.2),der(18)del(18)(q11.2q22)ins(18;13)(q11.2;q12q32)x2,der(21)del(21) 11p p11.2; ch11: (q11.2q22.3)ins(21;11)(?;p15p15),der(21)t(17;21)(q?;q21),der(22)t(16;22)(q22;p11.1), NE 151 +mar[cp22] 8 Ž/ M0 9 Ž / M2 42~43,X,-X,-3,der(4)t(4;10)(p12;p12),-5,der(10)t(10;17)(p12;q?)t(17;20)(?;?),der(11) t(5;11)(p12;p11.2),der(12)t(11;12)(p11.2;q13)inv(11)(p15.5p13)t(3;11)(?;p13)t(3;16) (?;q22),der(16)t(3;16)(?;q22),-17,der(17)t(12;17)(q13;q11.1),der(20)t(17;20)(q?;?) [cp22] 48~57,XX,+X,der(1)t(1;5)(q21.1;?),der(2)t(1;2)(q?;q33),der(5)t(5;17)(q11.2;?)t(5;18) (p13;?),+8,+8,+9,+10,+10,+der(11)t(1;11;21)(q?;p15;q?),der(12)t(12;20) (p12;?), +13,der(15)t(5;15;18)(q?;p11.1;?),-17,+18,+19,der(21)t(17;21)(?;p11.1),+der(22)t(1;22) (?;p11.1)x2[cp22] 11p p13 11p M/ M4 46,XY,t(11;20)(p11.2;q?)[16] 11p p15.5; ch11: p13; ch11: del(11)(p12p12) 11p12; ch11: p12; ch11: NE 78 11p15-11p15.4 (NUP98) NE 54 arr 11p13 ( )x1 11p15.4 (NUP98) 11p13; ch11: p13; ch11: p11.2; ch11: NE ~49,XY,+X,del(1)(q21),+r(1)(?),ins(5;1)(p15;?),del(11)(p12p13),der(12)t(1;12) 11p13 11p14.1; ch11: M/ saml (?;q12),ins(14;15)(q22;?),der(15)t(15;21)(q14;?)t(12;21)(q12;?),t(16;17)(?;p13),der(18) - NE 9 11p12 11p12; ch11: t(15;18)(q14;q21),+19[cp22] 44~45,X,-Y,der(1)t(1;5)(q12;?),-5,del(5)(q11),der(7)ins(7;5)(q11.2;?)t(1;7)(q12;q22), 12 M/ M2 11p14-11p13; ch11: NE ,der(11)t(11;16)(p14;?),der(12)t(5;12)(?;p13),-16,der(18)t(Y;18)(q11;p11)[cp19] 47,XY,der(2)t(2;15)(q34;q24),del(5)(q13q31),der(7)t(7;11)(q22;p13),del(11)(p13), 11p13 arr 11p11.2qter 11p12; ch11: M/ M1 NE 47 +idic(11)(p11.1),del(15)(q24),der(18)del(18)(p11.2)del(18)(q21)[cp22] 11p11.1 ( )x3 11p11.2; ch11: Ž/ sm0 45~48,XX,idic(5)(q11.1),dic(7;11)(q11.1;p11.1),+8,del(8)(q?),del(12)(p13),+13[cp22] NE žije Zkratky: M, muž; Ž, žena; s, sekundární; -, neprovedeno; alo-tkd, alogenní transplantace kostní dřeně; žije, stav k
40 Tabulka 6. Klinická data nemocných s abnormalitou chromosomu 11 a výsledky molekulárně cytogenetického mapování. Pacient č. Pohl/ Datum Typ Zlomové místo na chromosomu 11 + nově detekované změny alo- Přežití Souhrný karyotyp (Klasická cytogenetická analýza a/nebo mfish/mband) Věk diagnózy AML mband SNP array (Mb) FISH (Mb) TKD (dny) PŘESTAVBY GENU MLL 15 Ž/ M5a 47,XX,+8,t(9;11)(p22;q23)[14]/46,XX[2] q23.3 (MLL) NE 6 16 Ž/ M5a 0 mitos q23.3 (MLL) NE 6 17 Ž/ M5a 46,XX,t(7;11;10)(q22;q23;p12)[3] q23.3 (MLL) ANO žije 46,XX,der(10)t(8;10)(?;p12)[11]/48,XX,+9,del(9)(p?)x2,der(10)t(9;10)(p?;p12),+19 ANO 18 Ž/ M5a q23.3 (MLL) 241 [4]/46,XX[1] 19 Ž/ M5a 46,XX,t(9;12;11)(p22;p13;q23)[8]/46,XX[6] 11q q23.3 (MLL) ANO žije 20 Ž/ M5a 46,XX,t(9;11)(p22;q23)[2]/46,XX[19] q23.3 (MLL) NE Ž/ M1 45,XX,t(6;11)(q27;q23),-7[7]/46,XX[2] q23.3 (MLL) NE M/ M1 0 mitos q23.3 (MLL) ANO žije 23 Ž/ M1 46,XX,t(11;19)(q23;p13)[15]/46,XX[7] q23.3 (MLL) ANO Ž/ sm5a 46,XX,t(9;11)(p22;q23)[7]/46,XX[15] q23.3 (MLL) ANO žije 25 M/ sm4 46,XY,t(9;11)(p22;q23.3)[13]/46,XY[8] q23.3 (MLL) NE M/ M5 46~52,XY,t(2;11)(p24;q23.3),+4,+8,+8,+12,+16,+21[cp12] q23.3 (MLL) NE M/ M5 45~50,XY,+3,+8,t(9;11)(p22;q23),+der(9)t(9;11)(p22;q23),-13,+14[cp18] q23.3 (MLL) ANO žije 28 M/ sm1 46,XY,t(11;19)(q23;p13.3)[19]/46,XY[3] q23.3 (MLL) NE M/ sm5 46,XY,t(9;11)(p22;q23)[22] q23.3 (MLL) NE Ž/ M5a 46,XX,t(9;11)(p22;q23)[22] q23.3 (MLL) NE M/ M4 46,XY,t(9;11)(p22;q23)[22] q23.3 (MLL) NE M/ M1 46,XY,t(9;11)(p22;q23)[8]/46,XY[5] q23.3 (MLL) ANO žije 33 Ž/ sm4 46,XX,t(9;11)(p22;q23)[11]/46,XX[1] q23.3 (MLL) NE M/ M5a 46,XY,t(11;19)(q23;p13.3)[18]/46,XY[4] q23.3 (MLL) ANO žije 35 M/ M1 46,XY,t(11;19)(q23;p13.3)[18]/46,XY[3] q23.3 (MLL) NE Ž/ AML 46,XX,t(11;19)(q23;p13.1)[22] q23.3 (MLL) ANO Ž/ M5 46,XX,der(1)t(1;1)(p36.3;q12),t(9;11)(p22;q23)[cp18] q23.3 (MLL) ANO žije 38 Ž/ sm2 46,XX,t(11;17)(q23.3;q25)[26] q23.3 (MLL) NE M/ sm2 46,XY,t(1;11)(q21;q23.3)[3]/47,idem,+10[4]/46,XY[3] q23.3 (MLL) NE Ž/ sm1 92,XXXX,t(6;11)(p21.3;q23.3)x2[6] 11q23.3 arr(11)x2 11q23.3 (MLL) NE žije 41 M/ M5a 46,Y,ins(X;11)(q24;q23.3q13)[22] 42 M/ M4 40~42,XY,der(2)t(2;12)(q34;?)t(7;12)(p21;?),del(3)(p11p26),del(4)(q?),der(5)t(5;15) (q11.2;q25),del(6)(q23),-7,der(7)del(7)(p11)del(7)(q11),der(8)t(8;10)(q21;q24), der(11)t(7;11)(q31;p13),der(11)t(11;20)(q23.3;?),-12,t(13;21)(q12;q21),der(14) t(11;14)(q23.3;p11)t(1;11)(?;q23.3),der(15)t(11;15)(q23.3;q22),-16,-17,der(20) t(20;21)(q11;q21),-20,der(21)t(11;21)(q23.3;?),-22,t(22;22)(p11;q11)[cp29] Zkratky: M, muž; Ž, žena; s, sekundární; -, neprovedeno; alo-tkd, alogenní transplantace kostní dřeně; žije, stav k q13 11q p13 11q23.3 arr(11)x2 arr 11p13pter ( )x1 arr 11q14.3q21 ( )x1 arr 11q22.3q23.3 ( )x1 arr 11q23.3qter ( )x3 11q13.2 (ch11: ) 11q23.3 (MLL) 11p13; ch11: q14.2; ch11: q21; ch11: q22.3; ch11: q23.3; ch11: q23.3 (MLL) ANO žije NE 6
41 Tabulka 6. Klinická data nemocných s abnormalitou chromosomu 11 a výsledky molekulárně cytogenetického mapování. Pacient č. Pohl/ Datum Typ Zlomové místo na chromosomu 11 + nově detekované změny alo- Přežití Souhrný karyotyp (Klasická cytogenetická analýza a/nebo mfish/mband) Věk diagnózy AML mband SNP array (Mb) FISH (Mb) TKD (dny) OSTATNÍ ABNORMALITY DLOUHÉHO RAMENE CHROMOSOMU 11 (11q) 43 Ž/ M5b 46,XX,t(11;14)(q12;q31)[4] q12; nemapováno NE 7 44 Ž/ saml 45,XX,del(5)(q14q33),der(16)ins(16;9)(p13;?)t(16;19)(p13;?),-17,der(18)t(18;21) (p11;?)t(17;21)(q11;?)[6]/45,xx,del(5)(q14q33),der(11)inv dup(11)(q24q12) t(1;11)(p?;q12),-17,der(18)t(18;21)(p11;?)t(17;21)(q11;?)[5]/45,xx,del(5)(q14q33), der(16)t(7;16)(?;p13)t(7;20)(?;?)t(11;20)(q22;?),-17,der(18)t(18;21)(p11;?)t(17;21) (q11;?)[2] 49 M/72 20/07/2010 saml 46~51,X,der(Y)t(Y;10)(q11.2;q22),+8,+9,der(10)t(10;15)(q22;22),+13,+20,+21[15]/ 46,XY,der(11)hsr(11)(q14)[5]/46,XY[2] 50 Ž/ M2 44~45,XX,t(4;20)(q12;p11.2),del(5)(q13q33),der(7)t(7;17)(q21;q?),der(9)t(9;11) (q34;q22),+10,-11,-17[cp10] 51 Ž/ sm2 74~77<3n>,XXX,+1,-3,+4,+7,+8,+10,-11,ace(11)(q23.3q25)hsr(11)(q23.3)x1~3, del(11)(q23.3),+12,-16,+17,-19,+21,+22[cp5]/46,xx[3] Zkratky: M, muž; Ž, žena; s, sekundární; -, neprovedeno; alo-tkd, alogenní transplantace kostní dřeně; žije, stav k q12 11q22 11q24-11q12; nemapováno 11q13.2; ch11: q24; nemapováno 45 M/ M0 47,XY,+13,der(17)t(11;17)(q13;p11)[cp7]/46,XY[15] - arr 11q13.2qter ( )x3 11q13.2; ch11: NE M/ M4 46,XY,der(1)t(1;11)(p34.3;q14.3),+13[cp16]/46,XY[2] 11q q14.2; ch11: NE Ž/ saml 43,XX,-3,t(4;11)(q?;q22),der(5)t(5;16)(q12;?),+8,-9,der(14)t(14;20)(p10;?)t(3;20) arr 11q22.1q q21; ch11: (?;?),der(16)t(16;17)(?;q22),-17,-18,der(20)t(3;20)(?;?),der(22)t(9;22)(?;p10)[cp7]/ 11q22 ( )x1 delece 11q22.1 nepotvrzena 46,XX[3] NE Ž/ M0 44,XX,-3,del(5)(q13q33.1),der(7)t(3;7)(?;q31.3),der(11)hsr(q23.3),ins(12;11) (q13;q14q24)hsr(11)(q14),der(16)del(16)(p11)del(16)(q12),der(17)t(17;20)(p12;?), -20[cp16]/44,XX,-3,del(5)(q13q33.1),der(7)t(3;7)(?;q31.3),der(11)hsr(11)(q23.3) t(11;17)(q23;p12),der(16)del(16)(p11)del(16)(q12),-17[3] 11q14 11q q24 arr 11q21q22.3 ( )x3 arr 11q22.3.q24.1 ( )x4 arr 11q24.2q25 ( )x4 arr 11q25qter ( )x1 11q arr 11q23.1qter ( ) 11q21; ch11: q22.3; ch11: q24.1; nemapováno 11q24.2; nemapováno 11q25; nemapováno 11q21-q22.3; ch11: NE žije NE 22 NE 54 11q23.1; ch11: ANO q q23.3; ch11: NE 110
42 Tabulka 6. Klinická data nemocných s abnormalitou chromosomu 11 a výsledky molekulárně cytogenetického mapování. Pacient č. Pohl/ Věk Datum diagnózy Typ AML Souhrný karyotyp (Klasická cytogenetická analýza a/nebo mfish/mband) ABNORMALITY KRÁTKÉHO A SOUČASNĚ DLOUHÉHO RAMENE CHROMOSOMU M/ M2 44~45,XY,der(1)t(1;12)(q12;q13),der(3)t(3;18)(p12;q11),del(5)(q12q33),der(11) hsr(11)(p15)hsr(11)(p13)hsr(11)(q22.1),-12,der(17)t(1;17)(q12;p13),-18,+21[8]/ 44,XY,der(1)t(1;12)(q12;q13),der(3)t(3;18)(p12;q11),del(5)(q12q33),-7,der(9) ins(9;11)(p22;q22.1q23.3),r(11)(q?q?),-12,der(17)t(1;17)(q12;p13),-18,+21[cp4]/ 32,X,-Y,der(1)t(1;12)(q12;q13),-3,-5,-7,-10,-11,der(11)hsr(11)(p15)hsr(11)(p13) hsr(11)(q22.1)t(7;11)(q11.1;q23.3),-12,-13,-15,-16,der(17)t(1;17)(q12;p13),-18,-19, -20,-21,ins(22;11)(q12;q22.1q23.3)[3] 53 M/ M4 44,XY,t(3;7)(p?;?),der(11)(17?::21?::11p13 11q24::11p p15.1::11q q24::11p p15.1::11q q24::11p15.5 p11.2::9?),-17,-21[5]/ 46,XY[1] 54 M/ M2 46,XY,der(4)t(4;11)(q11;p15),der(5)t(5;11)(q35;q13),der(11)t(4;11)(q11;q13) t(5;11)(q35;p15)[21]/46,xy[1] Zkratky: M, muž; Ž, žena; s, sekundární; -, neprovedeno; alo-tkd, alogenní transplantace kostní dřeně; žije, stav k Zlomové místo na chromosomu 11 + nově detekované změny mband SNP array (Mb) FISH (Mb) 11p p p13 11p q q p p p13 11p q q24 11p15 11q13.5 arr 11p15.5pter (0-1.68)x1 arr 11p15.1p15.5 ( )x4 arr 11p12 ( )x3 arr 11q14.1q14.3 ( )x3 arr 11q14.3qter ( )x4 del(11)(p15.5pter)x1; amp(11)(11p15.5p15.1)x6; 11p15.5; ch11: p ; ch11: amp(11)(p13p12)x3; 11p13; ch11: p12; ch11: amp(11)(p12p12)x3; 11p12; ch11: p12; ch11: amp(11)(q14.1q14.2)x3; 11q14.1; ch11: q14.2; ch11: amp(11)(q14.2qter)x6; 11q14.2; ch11: alo- TKD Přežití (dny) NE NE 3-11p15.4 (NUP98) 11q13.2; ch11: NE 411
43 Tabulka 7. Přehled výsledků mapování zlomových míst na krátkém a dlohém rameni chromosomu 11. Číslo pacienta Oblast Použitá sonda Lokalizace der(11) der(11) der(11) der(21) der(11) +der(11) der(11) der(11) der(11) der(11) der(11) der(11) der(11) der(11) der(11) 11pter ToTel 11p RP11-348A p15.5 RP11-92N RP11-120E RP11-258P RP11-406D RP11-244O p14.1 RP11-242P RP11-258H RP4-562D RP11-140L RP11-366N RP-11-26B p13 RP11-74J RP11-98C RP11-68D RP11-514F RP11-465C RP11-233I RP11-144G RP11-160M RP11-40H p12 RP11-454H RP11-375D RP11-538D RP11-108L RP11-484D RP11-70A RP11-450F RP11-12C RP11-164L RP11-433B p11.2 RP11-425L RP11-368A RP11-165B RP11-169J RP11-390K RP11-17G RP11-125F Vysvětlivky:, přítomnost signálu;, duplikace;, amplifikace; nepřítomnost signálu;, zlom; -, neděláno; červeně ztráta oblasti; zeleně zisk oblasti; signál na derivovaném chromosomu umístěn telomericky; signál na derivovaném chromosomu umístěn centromericky.
44 Tabulka 7. Přehled výsledků mapování zlomových míst na krátkém a dlohém rameni chromosomu 11. Číslo pacienta Oblast Použitá sonda Lokalizace der(11) der(11) der(11) der(?) der(11) der(16) der(17) der(1) der(11) der(11) der(12) der(11) der(9) der(11) ace der(11) der(11) 11q13.1 RP11-472D RP11-424B RP11-715F RP11-98N q13.2 RP11-105D RP11-569N RP11-554A q13.3 LSI t(11;14) XT LSI CCND q13.4 RP11-92H RP11-139M RP11-15N q14.1 RP11-7H RP11-16H RP11-153F RP11-241M q14.2 RP11-496E q14.3 RP11-135L RP11-54L q21 RP11-159N q22.1 RP11-277H RP11-16K RP11-379J RP11-179B RP11-22J RP11-273B q22.3 LSI ATM RP11-268L RP11-18B q23.1 RP11-285P q23.2 RP11-267J RP11-215H q23.3 LSI MLL 11qter ToTel 11q Vysvětlivky:, přítomnost signálu;, duplikace;, amplifikace; nepřítomnost signálu;, zlom; -, neděláno; červeně ztráta oblasti; zeleně zisk oblasti; signál na derivovaném chromosomu umístěn telomericky; signál na derivovaném chromosomu umístěn centromericky.
45 Tabulka 8. Přehled zlomových míst identifikovaných na chromosomu 11 u AML. Oblast Frekvence Pacient č. Lokalizace (Mb) Kandidátní geny 11p15.5 n=5 6, 7, 8, 52, 53 ch11: cca 50 genů (HRAS, CD151, MUC6, MUC2, CARS) 11p15.4 n=3 9, 10, 54 ch11: NUP98 11p15.4-p15.1 n=3 7, 52, 53 ch11: cca 80 genů (RRM1, EIF3F, LMO1, HTATIP2, FANCF) 11p14.1 n=1 11 ch11: lincrna 11p13 n=7 11p12 11p11.2 n=7 n=5 53 nemapováno , 42 ch11: ch11: ch11: FSHB, KCNA4, C11orf46, MPPED2, DCDC5, DCDC1 DCDC5, DCDC1 DCDC5, DCDC1, DNAJC24, ELP4, IMMP1L, mirna 10 ch11: DNAJC24, ELP4, IMMP1L, mirna 52 ch11: TRAF6, RAG1, RAG2, C11orf74 13 ch11: Žádný protein/rna kódující gen 8 ch11: Žádný protein/rna kódující gen 52 ch11: RNA geny: SNORA31, U6, lincrna 52 ch11: LRRC4C 11 ch11: lincrna 8 52 ch11: ch11: lincrna Žádný protein/rna kódující gen 53 nemapováno 13 ch11: CD82, TP53I11, TSPAN18,lincRNA 10 ch11: DGKZ, MDK, CHRM4, AMBRA1 7 ch11: ARFGAP2, PACSIN3 7 ch11: SPI1, MYBPC3, MADD 11p11.1 n=1 14 nemapováno 11q12 n=2 43, 44 nemapováno 11q13.2 n= ch11: ch11: , 54 ch11: GAL, MTL5, lincrna 11q14.1 n=1 52 ch11: ODZ4 52 ch11: q ch11: n=5 52 ch11: q21 n=4 11q22.1 n=1 53 nemapováno 42 11q22.3 n=2 48 NDUFS8, UNC93B1, ALDH3B1, TCIRG1, CHKA, ALDH3B2,lincRNA, mirna, U6 cca 25 genů (ODZ4, RSF1, THRSP, GAB2, PICALM, MIR708, RAB30, SYTL2, EED, FZD4) cca 20 genů (RAB38, TYR, NOX4, TRIM49, NAALAD2, CHORDC1, FAT3) 42 ch11: cca 30 genů (FAT3, MRE11A, MTNRIB, PANX1, FUT4) 48 ch11: KDM4D, ENDOD1, CWC15, SESN3, KDM4DL ch11: ch11: ch11: CEP57, FAM76B, MTMR2, 5S RNA, lincrna cca 30 genů (MAML2) MAML2 ch11: ch11: GRIA4, U6, U4, KIAA1826, KBTBD3, AASDHPPT, lincrna cca 11 genů (GUCY1A2, ALKBHB, RAB39A, CUL5, ACAT1, NPAT) 11q23.1 n=1 50 ch11: ARGHAP20, RDX, ZC3H12C, FDX1 51 ch11: q ch11: n=31 42 ch11: q24 n=3 cca 30 genů (POU2AF1, MIR34B, MIR34C, CRYAB, SIK2) cca 30 genů (ZBTB16, NNMT, TAGLN, APOA4, APOC3) cca 10 genů (TMPRSS4, UBE4A, AMICA1, MPZL2) ch11: MLL 44 nemapováno 48 ch11: TECTA, SC5DL, SORL1 48 ch11: cca 25 genů (SPA17, ESAM, ROBO3, ROBO4, HEPACAM) 11q25 n=1 48 ch11: OPCML, NTM, C11orf39 Vysvětlivky: n, počet; lincrna, dlouhé nekódující RNA, rekurentní zlom, potenciálně nenáhodný, překrývající se oblasti.
46 Tabulka 9. Detekované oblasti nebalancovaných a balancovaných změn na chromosomu 11 u AML (a,b, 2 změny o různé velikosti v 1 klonu nebo 2 různé klony (viz text)). Změna Oblast Proxim./ distál. zlom(mb) Počet Číslo pacienta Počet genů Kandidátní geny z alterované oblasti 11p15.5 p n=1 7 > 80 RPLP2, NUP98 (nukleární transport), HRAS, CD151, CARS (trna syntéza) 11p15.4 p n=2 7, 9 > 80 5 NUP98 (nukleární transport), LMO1, RPL27A, EIF4G2 (iniciace translace), RPS13, FANCF (chromosomová nestabilita) 11p14.1 p n=1 9 > 100 DCDC5 (polymerace mikrotubulínu), DCDC1 (polymerace mikrotubulínu), DNAJC24 (aktivace Hsp70 chaperonů), IMMP1L (mitochondriální peptidáza), WT1, LMO2 (regulátor hematopoézy), RAG1, RAG2 (rekombinace), TRAF6, API5 (inhibitor apoptózy), CREB3L1, MDK, DDB2 (reparace DNA), MADD (regulace přežití), SPI1, SNORA31, U6 RNA, mnoho mirna a lincrna 11p11.1 q n=2 9, 14 > 100 PTPRJ, CTNND1, FEN1 (endonukleáza), mnoho mirna a lincrna 11q12 q n=3 9, 14, 44a > 100 MACROD1, BAD (inhibitor apoptózy), RELA, mnoho mirna a lincrna 11q13.1 q n=4 9, 14, 44a, 45 > 100 RAD9A (rekombinace), CHKA (metabolismus), MTL5 (buněčný růst), THRSP (metabolismus), GAB2, Duplikace mnoho mirna a lincrna 11q14.1 q n=5 9, 14, 44a, 45, 46 > 30 ODZ4, PICALM, PICALM, RAB30, EED (remodelace chromatinu), FZD4, mnoho mirna a lincrna 11q14.3 q n=6 9, 14, 44a, 44b, 45, 46 cca 30 FAT3, PANX1, množství malých jaderných RNA 11q21 q n=7 9, 14, 44a, 44b, 45, 46, 48a > 50 MRE11A (rekombinace), FUT4 (fukosyltransferáza), NOX4 (metabolismus), RAB38, MAML2, CEP57 (součást centrosomu, translokin), BIRC3, BIRC2 (inhibitor apoptózy), CASP1, CASP4, CASP5, CASP12 (kaspázy), GUCY1A2, CUL5 (ubiquitinylace), RAB39, NPAT (přechod z G1 do S fáze), DDX10 (RNA helikáza), mnoho mirna a lincrna 11q22.3 q n=8 9, 14, 44a, 44b, 45, 46, 48a, 50 > 80 ARHGAP20, POU2AF1, NNMT (metyltransferáza), TMPRSS4 (serinová proteáza), RAB39, ZBTB16, BTG4 (inhibitor apoptózy), SIK2, 5 MLL, MIR34B, MIR34C 11q23.3 q n=9 9, 14, 27, 44a, 44b, 45, 46, 48a, 50 > 50 3 MLL, DDX6 (RNA helikáza), H2AFX (histon), ARHGEF12, MIR100, SPA17, mnoho mirna a lincrna 11q24 qter n=7 9, 14, 27, 44b, 45, 46, 50 > 50 ETS1 (inhibitor apoptózy), NFRKB, U13 RNA, mnoho mirna a lincrna 11p15.5 p n=2 52, 53 > 150 NUP98 (nukleární transport), HRAS, CD151, LMO1, RPL27A, EIF4G2 (iniciace translace), RPS13, FANCF (chromosomová nestabilita) 11p n=1 53 > 30 PAX6, WT1, LMO2 (regulátor hematopoesy), CD44, mnoho mirna a lincrna 11p13 p n=2 52, 53 cca 30 RAG1, RAG2 (rekombinace), API5 (inhibitor apoptózy), TRAF6, SNORA31, U6 RNA, mnoho mirna a lincrna 11p12 p n=2 13, 53 cca 40 CREB3L1, MDK, DDB2 (reparace DNA), MADD (regulace přežití), SPI1, mnoho mirna a lincrna 11q14.1 q n=2 13, 52 > 50 GAB2, ODZ4, THRSP (metabolismus), PICALM, RAB30, EED (remodelace chromatinu), FZD4, MRE11A (rekombinace), FUT4 (fukosyltransferáza), mnoho mirna a lincrna 11q22.1 q n=4 13, 49, 52, 53 cca 40 MAML2, CEP57 (součást centrosomu, translokin), BIRC3, BIRC2 (inhibitor apoptózy), CASP1, CASP4, Amplifikace CASP5, CASP12 (kaspázy) 11q22.3 q n=5 13, 48b, 49, 52, 53 cca 15 GUCY1A2, CUL5 (ubiquitinylace), RAB39, NPAT (přechod z G1 do S fáze), DDX10 (RNA helikáza), mnoho mirna a lincrna 11q23.1 q n=6 13, 48b, 49, 51, 52, 53 > 80 ARHGAP20, POU2AF1, NNMT (metyltransferáza), TMPRSS4 (serinová proteáza), RAB39, ZBTB16, BTG4 (inhibitor apoptózy), SIK2, MIR34B, MIR34C 11q n=7 13, 42, 48b, 49, 51, 52, 53 cca 10 UBE4A (ubiquitinylace) 11q n=8 13, 18, 42, 48b, 49, 51, 52, MLL 11q23.3 q n=7 13, 42, 48b, 49, 51, 52, 53 > 80 3 MLL, DDX6 (RNA helikáza), H2AFX (histon), ARHGEF12, MIR100, SPA17, ETS1 (inhibitor apoptózy), NFRKB, mnoho mirna a lincrna 11q25 qter n=2 13, 42 cca 20 U13 RNA, mirna, lincrna Vysvětlivky: transkripční faktor, tumor supresor, signalizace, ribozomální protein či RNA, RNA pro úpravy mrna, ostatní nekódující RNA, jiné.
47 Tabulka 9. Detekované oblasti nebalancovaných a balancovaných změn na chromosomu 11 u AML (a,b, 2 změny o různé velikosti v 1 klonu nebo 2 různé klony (viz text)). Změna Oblast Proximální/ distální zlom (Mb) Počet Číslo pacienta Počet genů Kandidátní geny z alterované oblasti 11pter p n=4 8, 12, 42, 52 > 80 MIR210 (inhibice proliferace), RPLP2, HRAS, CD151, MUC6, MUC2, MUC5B (mucíny, ochrana povrchu žaludku, nádorový marker), CDKN1C (p57, inhibitor cyklin dependentní kinázy), CARS (trna syntéza) 11p15.5 p n=3 7, 12, 42 > 80 RRM1 (reparace DNA), EIF3F (inhibice translace), RPL27A, ST5 (funkce neznámá), EIF4G2 (iniciace translace), RPS13, HTATIP2 (iniciace apoptózy), FANCF (chromosomová nestabilita) 11p14.1 p n=4 7, 11, 12, 42 cca 24 MPPED2 (inhibice proliferace), DCDC5 (polymerace mikrotubulínu), 12x lincrna, 3x mirna 11p n=4 7, 10, 11, 42 7 DCDC1 (polymerace mikrotubulínu), DNAJC24 (aktivace Hsp70 chaperonů), IMMP1L (mitochondriální peptidáza), 3x mirna Delece 11p n=3 7, 11, 42 4 ELP4 (remodelace chromatinu), RCN1 (Ca 2+ vazebný protein), PAX6 11p13 p n=2 7, 11 > 30 WT1, LMO2 (regulátor hematopoesy), RAG1, RAG2 (rekombinace), TRAF6, CD44 11p n=3 7, 8, LRRC4 (regulace axenogeneze), SNORA31, U6 RNA, 4x lincrna, 5x mirna 11p n=2 7, 8 2 2x lincrna 11p12 p n=1 7 cca 40 API5 (inhibitor apoptózy), CD82 (suprese metastáz), CREB3L1, DGKZ (regulace kinázy C), MDK, DDB2 (reparace DNA), MADD (regulace přežití), SPI1 11q14.3 q n=1 42 > 20 FAT3, MRE11A (rekombinace), FUT4 (fukosyltransferáza) 11q22.3 q n=1 42 >50 ATM, ARHGAP20, POU2AF1, NNMT (metyltransferáza), TMPRSS4 (serinová proteáza), RAB39, ZBTB16, MIR34B, MIR34C 11q25 qter n=2 44a, OPCML, NTM (adhezivní molekula), U6 RNA, U13 RNA, 3x mirna, 5x lincrna 11p n=1 6 cca 50 MIR210 (inhibice proliferace), RPLP2, HRAS, CD151, MUC6, MUC2, MUC5B (mucíny, ochrana povrchu žaludku, nádorový marker), CDKN1C (p57, inhibitor cyklin dependentní kinázy), CARS (trna syntéza) 11p n=2 10, 54 1 NUP98 (nukleární transport) 11p n=1 8 8 DCDC5 (polymerace mikrotubulínu), DCDC1 (polymerace mikrotubulínu), DNAJC24 (stimulace ATPáz), ELP4 (remodelace chromatinu), IMMP1L (mitochondriální peptidáza), 3x mirna Balancovaná 11p n=1 13 Žádný protein/rna kódující gen přestavba 11p n= DGKZ, MDK (růstový faktor), CHRM4 (inhibitor adenylát cyklázy), AMBRA1 (vývoj nervového systému), mirna 11p n=1 7 3 SPI1, MYBPC3 (myozin vazebný protein), MADD 11q12?? n= q n=2 41, 54 4 GAL (galanin, neuropetid), MTL5 (buněčný růst a diferenciace), lincrna, mirna 11q n= CEP57 (součást centrosomu, translokin), FAM76B (funkce neznámá), MTMR2 (metabolismus), 5S RNA, 2x lincrna, mirna 11q n= , 19-26, MLL Vysvětlivky: transkripční faktor, tumor supresor, signalizace, ribozomální protein či RNA, RNA pro úpravy mrna, ostatní nekódující RNA, jiné. 47
 a delece (červeně) na chromosomu 11 u AML")
48 Obrázek 8. Detekované oblasti duplikace/amplifikace (zeleně) a delece (červeně) na chromosomu 11 u AML (a,b, dvě změny o různé velikosti v jednom klonu nebo dva různé klony (viz text)).
49 Charakteristika a klinický význam zlomových míst Změny 11p Alteraci krátkého ramene chromosomu 11 jsme prokázali u 13 z 54 (24 %) nemocných (viz tabulka 6). Porovnání klinických a cytogenetických dat nemocných dle postižení ramene chromosomu je zobrazeno v tabulce 16. Všechny alterace 11p byly součástí komplexního karyotypu, často detekovány spolu s dalším zlomem na chromosomu 11. Mechanizmy vzniku zlomových míst byly: balancované přestavby (n=6; nemocní č. 6, 7, 8, 10, 13, 54), delece (n=6; nemocní č. 7, 8, 10, 11, 12 a 42) a duplikace/amplifikace (n=5; nemocní č. 9, 13, 14, 52, 53). Pomocí FISH mapování jsme na 11p identifikovali 18 různých zlomových míst. Rekurentní zlom byl identifikován v oblasti 11p15.4 v genu NUP98 a další potenciálně nenáhodné zlomy v pruzích 11p13 (ch11: Mb) a 11p12 (ch11: Mb) (viz tabulka 9). Opakované alterace jsme prokázali i v regionech 11p15.5 a 11p15.4-p15.1. Ty ale nebyly z důvodu nedostatku materiálu přesněji specifikovány. Ostatní zlomy se vyskytovaly jen sporadicky. Přestavbu genu NUP98 jsme detekovali u tří nemocných (č. 9, 10 a 54, viz tabulka 6 a 17). Ve všech třech případech byly kromě alterace NUP98 přítomné další změny chromosomů (u pacientů č. 10 a 54 i další zlomy na stejném chromosomu 11). U nemocných č. 10 a 54 měla přestavba genu NUP98 reciprokou povahu. Jednalo se o variantní komplexní translokaci zahrnující tři chromosomy: t(5;11;20)(q35;p15;p?) (nemocný č. 10) a t(4;5;11)(q11;q35;p15) (nemocný č. 54). U obou pacientů jsme pomocí FISH s BAC sondou RP11-265K23 prokázali fúzi genu NUP98 s genem NSD1 na 5q35. U nemocného č. 10 byla tato přestavba odhalena pouze metodou FISH. U nemocné č. 9 měla alterace genu NUP98 nebalancovanou povahu. Vznikla přestavbou nadpočetného chromosomu 11, jehož telomerická část krátkých ramen 11p pter byla nahrazena genetickým materiálem z chromosomu 1. Ve výsledku se tak jednalo o parciální trisomii 11 oblasti 11p15.4 (zahrnující pouze 5 konec NUP98) až 11qter. Ve FISH analýze jsme sledovali 3 kopie sondy RP11-258P13 značící 5 konec NUP98 a pouze 2 kopie sondy RP11-92N11 značící 3 konec NUP98. Ztráta oblasti 11p pter nadpočetného chromosomu byla potvrzena také subtelomerickou sondou pro 11p. U čtyř nemocných (č. 8, 10, 12, 42) jsme zlomové místo v oblasti 11p13 specifikovali do společného regionu ch11: Mb (viz tabulka 6 a 17). U
50 dvou pacientů (č. 8 a 10) se na stejném derivovaném chromosomu 11 nacházeli i další zlomy. Ve dvou případech (č. 10 a 42) byl aberacemi postižen i druhý homologní chromosom 11. U nemocné č. 8 došlo k zlomu při translokaci invertované části chromosomu 11 do derivovaného chromosomu 12. Jednalo se tedy o balancovanou změnu. U nemocného č. 10 vznikl zlom s následkem kryptické intersticiální delece v oblasti 11p13. Také v případě pacientů č. 12 a 42 měla změna nebalancovaný charakter. U obou se jednalo o nereciprokou translokaci vedoucí k terminální ztrátě krátkého ramene 11p13 11pter. Zlom v oblasti 11p12 v regionu ch11: Mb jsme identifikovali u nemocných č. 8 a 13 (viz tabulka 6 a 17). U nemocné č. 8 vznikl zlom na základě kryptické delece v oblasti 11p12, u pacienta č. 13 nereciprokou translokací 11p12 11pter na derivovaný chromosom 7. Při přestavbě nedošlo k ztrátě ani zisku genetického materiálu z chromosomu 11, proto byl zlom zařazen do skupiny balancovaných změn chromosomu 11. Změny 11q Alteraci dlouhého ramene chromosomu 11 jsme prokázali u 40 pacientů (č ), u čtyř současně s abnormalitami 11p (č. 42, 52, 53, 54). Protože literatura doporučuje odlišovat přestavby MLL genu jako samostatný specifický soubor nemocných, jsou změny 11q dále rozděleny na přestavby MLL genu a ostatní změny 11q (porovnání klinických a cytogenetických dat nemocných dle postižení ramene chromosomu je zobrazeno v tabulce 16). Změny genu MLL Přestavbu MLL genu jsme pozorovali u 28 z 54 pacientů (52 %). Identifikovali jsme tři nebalancované přestavby MLL genu (nemocní č. 18, 27 a 42) spojené se ziskem 5 nebo 3 konce MLL a 25 balancovaných (viz tabulka 6 a 17). Translokace MLL genu byla u 15 nemocných samostatnou aberací, u čtyř (č. 15, 20, 21, 39) byla detekována spolu s početní odchylkou jiného chromosomu (+8 nebo -7), u dvou šlo o komplexní translokaci/inzerci s celkem třemi zlomovými místy na účastněných chromosomech (č. 17, 19 a 41), a u pěti (č. 18, 26, 27, 37, 42) se vyskytovala s dalšími početními/strukturními přestavbami. V sedmi případech byla přestavba MLL genu prokázána pouze metodou FISH. Zatímco u dvou nemocných nebyly přítomné dělící se buňky (č. 16 a 22), u tří pacientů se změnu genu nepodařilo zachytit klasickou 50
51 cytogenetickou analýzou: t(9;11)(p22;q23) (fúzní gen MLL-MLLT3) u nemocného č. 15, t(9;12;11)(p22;p13;q23) (fúzní gen MLL-MLLT3) u nemocného č. 19 a t(6;11)(q27;q23) (fúzní gen MLL-MLLT4) u nemocného č. 21. Ve dvou případech (č. 18, 26) došlo ke kryptické inserci MLL genu do derivovaného chromosomu 10 (fúzní gen MLL-MLLT10) v rámci rozsáhlých změn karyotypu. U obou nemocných se aberaci nepodařilo odhalit ani metodou mfish, přítomnost 5 konce MLL genu na 10p12 tak byla prokázána pouze kombinací translokační sondy pro MLL gen a centromerické sondy pro chromosom 10. U nemocného č. 26 byla inserce maskována zdánlivě reciprokou translokací t(2;11)(p24;q23). Nejčastějšími přestavbami MLL genu byly t(9;11)(p22;q23) (n=13; jednou při nálezu 0 mitos, jednou v rámci variantní translokace), t(11;19)(q23;p13) (n=5) a translokace/inzerce (10;11)(p12;q23) (n=3; jednou v rámci variantní translokace a dvakrát kryptická spolu s přídatnými aberacemi dalších chromosomů). U jednoho nemocného jsme identifikovali novou, dosud nepublikovanou, translokaci MLL genu t(6;11)(p21.3;q23) v tetraploidním karyotypu. Zlomové místo na partnerském chromosomu 6 bylo lokalizováno centromericky od oblasti 6p21.31 hybridizující k BAC sondě RP11-174A4. Ostatní změny 11q Změnu dlouhého ramene chromosomu 11, jejíž součástí nebyla přestavba genu MLL, jsme prokázali u 14 pacientů (č ), u tří současně s abnormalitami krátkého ramene (č. 52, 53, 54). Porovnání klinických a cytogenetických dat nemocných dle postižení ramene chromosomu je zobrazeno v tabulce 16. U desíti nemocných byly alterace 11q součástí komplexního karyotypu (č. 42, 44, 47-54). Mechanizmy vzniku zlomových míst na 11q byly: balancovaná přestavba (nemocní č. 41, 43, 47, 54), duplikace/amplifikace (nemocní č. 42, 44, 45, 46, 48 53) a delece (č. 42, 44, 48). Pomocí FISH mapování jsme na 11q kromě genu MLL identifikovali 21 dalších různých zlomových míst. Potenciálně nenáhodný zlom se vyskytl v pruhu 11q13.2 ( Mb) (viz tabulka 7). Opakované alterace jsme prokázali i v regionech 11q12, 11q13.2, 11q14 a 11q24. Z důvodu nedostatku materiálu ale nebyly tyto zlomy více specifikovány. Ostatní zlomy se vyskytly jen sporadicky. Zlom v 11q13.2 regionu ch11: Mb jsme detekovali u dvou mužů (viz tabulka 6 a 17). U obou nemocných vznikl zlom na základě balancované přestavby a také se na stejném derivovaném chromosomu 11 nacházel i další zlom (v genu NUP98 u pacienta č. 54 a v genu MLL u pacienta č. 41). 51
52 Změny 11p i q Změnu dlouhého a současně krátkého ramene chromosomu 11 jsme prokázali u čtyř pacientů (č. 42, 52, 53, 54) (viz tabulka 6). U nemocných č. 42, 52 a 53 vznikla zlomová místa na obou ramenech vlivem nebalancovaných změn způsobených amplifikacemi (č. 42, 52, 53) nebo delecemi (č. 42). Balancovanou tří cestou translokaci se dvěmi zlomovými místy na chromosomu 11 (na 11p v genu NUP98) jsme popsali u pacienta č. 54. Parciální tandémové a netandémové duplikace MLL genu Pomocí molekulárně genetických metod jsme parciální tandémovou duplikaci genu MLL detekovali u 50 (23 %) nemocných a parciální netandemovou duplikaci MLL u tří (1 %) nemocných. Soubor nemocných s MLL PTD tvořilo 23 žen a 27 mužů, medián věku 55 let. V devíti případech se jednalo o sekundární formu AML. FAB subtyp byl stanoven jako: M2 (n=18), M1 (n=13), M4 (n=10), M0 (n=4), M5 (n=2), M6 (n=2), jiný (n=1). Dle nálezu cytogenetické analýzy se MLL PTD nacházela spolu s: normálním karyotypem (n=21), nálezem 0 mitos (n=3), prognosticky příznivou změnou t(8;21)/inv(16) (n=4), komplexním karyotypem (n=5, z toho 2x s aberací chromosomu 11 a 1x s trisomií 11), přestavbou MLL genu (n=7), s dalšími chromosomovými abnormalitami (n=10, z toho 1x s parciální trisomií 11, 1x s úplnou trisomií 11, 2x s monosomií 7). Celkem se MLL PTD vyskytovala současně s nepřerušenou přestavbou MLL genu u 7 nemocných, s jinou změnou chromosomu 11 u čtyřech nemocných (viz tabulka 10). Při MLL PTD došlo nejčastěji k duplikaci exonů 2 až 6 (29x), dále exonů 2 až 8 (8x) a 2 až 7 (3x). U devíti nemocných byla duplikace přerušena delecí nukleotidů v různých exonech o různé velikosti. Neparciální tandemovou duplikaci MLL genu jsme identifikovali u tří žen (medián věku 43 let). Všechny měly primární AML typu M5 (n=2) a M1 (n=1). MLL PNTD se vyskytovala společně s translokacemi t(9;11)(p22;q23), t(11;19)(p13;q23) a t(9;12;11)(p22;p13;q23) (viz tabulka 11, obrázek 9). U pacienta č. 20 byla navíc prokázána delece v exonu 7 genu MLLT3 (ztráta 21 nukleotidů) a v exonu 1 genu MLL (ztráta 438 nukleotidů). 52
53 Tabulka 10. Přehled MLL PTD u nemocných se změnou chromosomu 11. Pacient Změna chromosomu 11 Duplikované MLL exony 1 trisomie trisomie 11 v rámci KK přestavba 11p v rámci KK přestavba MLL genu - t(6;11)(q27;q23) přestavba MLL genu - t(9;11)(p22;q23) přestavba MLL genu - t(9;11)(p22;q23) přestavba MLL genu - t(9;11)(p22;q23) přestavba MLL genu - t(9;11)(p22;q23) přestavba MLL genu - t(11;19)(q23;p13.3) přestavba MLL genu - t(9;11)(p22;q23) parciální trisomie 11q balancovaná přestavba 11p i 11q 2 6 Vysvětlivky: KK, komplexní karyotyp. Tabulka 11. Přehled MLL PNTD u nemocných se změnou chromosomu 11. Pacient Přestavba MLL genu Duplikace MLL exonů Partnerský gen (PG) Exony PG přerušující MLL PTD 19 t(9;12;11)(p22; p13;q23) 2 8 MLLT3 9,10 20 t(9;11)(p22;q23) 1 10 MLLT3 6,7 23 t(11;19)(q23;p13.3) 2 8 MLLT1 2,3 Obrázek 9. Struktura MLL PNTD (A, pacient č. 19; B, pacient č. 23). A) B) 53
54 Porovnání doby přežití nemocných s AML se změnou chromosomu 11 Na 5% hladině jsme prokázali statisticky významný rozdíl mezi dobou přežití nemocných se: 1) změnou 11p, přestavbou MLL genu a ostatními změnami 11q (viz tabulka 12 a graf 3). 2) duplikací MLL genu, amplifikací MLL genu a balancovanou přestavbou MLL genu (viz tabulka 13 a graf 4). Na 0,1% hladině jsme prokázali statisticky významný rozdíl mezi dobou přežití nemocných s: 1) normálním karyotypem, přestavbou MLL genu a ostatními změnami chromosomu 11 (tabulka 14, graf 5). 2) normálním karyotypem, změnami chromosomu 11 včetně přestaveb MLL genu a komplexního karyotypu, jehož součástí byl/nebyl chromosom 11 (tabulka 15, graf 6). Statisticky významný rozdíl se nepodařilo prokázat v době přežítí nemocných se: 1) ziskem/ztrátou a balancovanou změnou chromosomu 11. 2) balancovanou přestavbou MLL genu a dalšími balancovanými přestavbami chromosomu 11. 3) balancovanou a nebalancovanou přestavbou genu MLL. 4) translokacemi t(9;11), t(11;19), ins(10;11) a ostatními přestavbami MLL. 5) normálním karyotypem a přestavbou genu MLL. 6) přestavbou genu MLL samostatnou či s jednou početní odchylkou a přestavbami MLL genu s dalšími strukturními nebo početními aberacemi. 7) přestavbami MLL a MLL PTD+, přestavbami MLL a MLL PTD- a MLL PNTD. 8) normálním karyotypem s MLL PTD+ a normálním karyotypem s MLL PTD- 54
55 Nález Tabulka 11. Přehled průměrné délky nemocných s jednotlivými nálezy zavzaté do statistické analýzy pro porovnání křivek přežití. n žíjící průměr (dny) medián (dny) n % odhad chyba odhad chyba Normální karyotyp, MLL PTD ,1% 720,39 92,96 495,00 114,18 Normální karyotyp, MLL PTD ,6% 1072,80 174, ,00 407,37 Přestavba MLL, MLL PTD ,9% 690,38 217,85 429,00 60,70 Přestavba MLL, MLL PTD ,7% 943,13 299,86 162,00 198,40 MLL PNTD ,3% 793,67 466,19 369,00 229,44 Přestavba MLL + 1 početní odchylka ,0% 446,74 117,15 202,00 187,48 Přestavba MLL + další přídatné změny ,5% 1396,38 357,48.. t(9;11)(p22;q23) ,2% 955,85 249,82 371,00. t(11;19)(q23;p13) ,0% 357,80 80,86 406,00 40,53 ins(10;11)(p12;q23) ,3% 817,67 557,49 241,00 169,02 Ostatní translokace MLL ,3% 354,42 159,16 154,00 144,03 Duplikace MLL ,0% 353,40 170,76 89,00 94,08 Amplifikace MLL 6 0 0% 50,17 15,10 47,00 19,60 Přestavba MLL ,7% 846,20 185,91 339,00 106,64 Nebalancovaná přestavba MLL ,3% 403,67 235,37 241,00 191,88 Balancovaná přestavba MLL ,0% 847,01 197,02 369,00 132,06 Balancovaná změna chromosomu 11 bez MLL ,0% 238,00 109,50 129,00 136,67 Změny chromosomu 11 bez přídatných změn ,3% 838,45 168,93 241,00 138,53 Komplexní karyotyp (s chromosomem 11) ,3% 138,84 33,84 78,00 40,63 Komplexní karyotyp (bez chromosomu 11) ,0% 293,14 93,82 114,00 36,44 Změny 11p ,1% 120,55 35,59 78,00 35,78 Změny 11p+q 4 0 0% 121,25 97,63 6,00 31,00 Změny 11q bez účasti MLL 8 0 0% 130,87 51,30 89,00 39,60 Změny 11q s účastí MLL ,6% 700,93 156,07 202,00 107,96 Duplikace/amplifikace chromosomu ,8% 354,77 135,83 89,00 45,00 Delece chromosomu % 121,60 36,87 142,00 70,11 Balancovaná změna chromosomu ,5% 791,65 181,57 369,00 137,09 Trisomie chromosomu ,0% 650,40 310,26 193,00 164,32 Parciální trisomie chromosomu ,7% 293,83 136,10 89,00 72,87 Amplifikace chromosomu % 94,60 40,36 65,00 12,05 55

56 Tabulka 12a. Porovnání přežití u nemocných se změnou 11p, přestavbou MLL genu a ostatními změnami 11q. Nález n exit žije n procento Změny 11p ,1% Ostatní změny 11q 8 8 0,0% MLL přestavby ,6% Celkem ,3% Tabulka 12b. Průměrná doba a medián přežívání ve dnech. průměr medián odhad chyba odhad chyba Změny 11p 120,56 35,58 78,00 35,78 Ostatní změny 11q 130,87 51,30 89,00 39,60 MLL přestavby 823,70 190,82 339,00 102,44 Celkem 568,41 132,06 154,00 28,84 Mantel-Cox test (funkcí přežívání) = 7,574 p = 0,023 (0,05) Graf 3. Funkce přežívání u nemocných se změnou 11p, přestavbou MLL genu a ostatními změnami 11q. 56
57 Tabulka 13a. Porovnání přežití u nemocných s duplikací MLL genu, amplifikací a balancovanou přestavbou. Nález n exit žije n procento Duplikace MLL ,0% Amplifikace MLL 6 6 0,0% Balancovaná přestavba MLL ,0% Celkem ,8% Tabulka 13b. Průměrná doba a medián přežívání ve dnech. průměr medián odhad chyba odhad chyba Duplikace MLL 353,40 170,76 89,00 94,08 Amplifikace MLL 50,17 15,10 47,00 19,60 Balancovaná přestavba MLL 847,01 197,02 369,00 132,06 Celkem 650,00 143,17 162,00 66,57 Mantel-Cox test (funkcí přežívání) = 8,675 p = 0,013 (0,05) Graf 4. Funkce přežívání u nemocných s duplikací MLL genu, amplifikací a balancovanou přestavbou. 57

58 Tabulka 14a. Porovnání přežití u nemocných s normálním karyotypem, přestavbou MLL a ostatními změnami chromosomu 11. Nález n exit žije n procento Normální karyotyp ,1% Přestavby MLL ,7% Ostatní změny chromosomu ,0% celkem ,6% Tabulka 14b. Průměrná doba a medián přežívání ve dnech. průměr medián odhad chyba odhad chyba Normální karyotyp 720,39 92,96 495,00 114,18 Přestavby MLL 846,20 185,91 339,00 106,64 Ostatní změny chromosomu ,11 86,07 89,00 37,47 celkem 727,07 90,72 329,00 60,62 Mantel-Cox test (funkcí přežívání) = 17,163 p = 0,001 (0,001) Graf 5. Funkce přežívání u nemocných s normálním karyotypem, přestavbou MLL genu a ostatními změnami chromosomu
 19 18 1 5,3% KK (ostatní) 25 20 5 20,0% celkem 135 93 42 31,1% Tabulka 15b.")
59 Tabulka 15a. Porovnání přežití u nemocných s normálním karyotypem, změnami chromosomu 11 bez mnohočetných přídatných chromosomových změn a komplexním karyotypem (KK) s/bez účasti chromosomu 11. Nález n exit n žije procento Normální karyotyp ,1% Změny chromosomu ,3% KK (součástí chromosom 11) ,3% KK (ostatní) ,0% celkem ,1% Tabulka 15b. Průměrná doba a medián přežívání ve dnech. průměr medián odhad chyba odhad chyba Normální karyotyp 720,39 92,96 495,00 114,18 Změny chromosomu ,45 168,93 241,00 138,53 KK (součástí chromosom 11) 138,84 33,84 78,00 40,63 KK (ostatní) 293,14 93,82 114,00 36,44 celkem 671,62 80,17 241,00 49,25 Mantel-Cox test (funkcí přežívání) = 25,509 p = 0,001 (0,001) Graf 6. Funkce přežívání u nemocných s normálním karyotypem, změnami chromosomu 11 a KK, jehož součástí je/není chromosom
60 Tabulka 16. Porovnání klinických a cytogenetických dat u nemocných dle postižení ramene chromosomu. Postižené rameno 11p 11q (MLL gen) 11q (ostatní) Rekurentní zlomy Frekvence* 11p15.4 (NUP98) 11p13 11p12 13/24 % (4x s 11q) 11q23.3 (MLL) 28/52 % (1x s 11p) 11q /26 % (3x s 11p) Ø věk Poměr Ž/M Poměr P/S FAB typ (počet) 61 5/8 9/4 M2(4) M4(4) M0(2) M1(1) M6(1) jiné(1) 46 15/13 20/8 M5(14) M1(7) M4(4) M2(2) jiné(1) 62 6/8 10/4 M2(4) M4(3) M0(2) M5(2) Mechanismus zlomu (počet) BP(6) delece(6) duplikace/ amplifikace(5) Komplexní Dosažení alo- žije Ø doba přežití karyotyp 1. remise TKD (měsíce) jiné(3) Vysvětlivky: * % z počtu nemocných s abnormalitou chromosomu 11; Ž, ženy; M, muži, P, primární; S, sekundární; BP, balancovaná přestavba; NP, nebalancovaná přestavba; alo-tkd, alogenní transplantace kostní dřeně. BP(25) NP(3) BP(4) duplikace/ amplifikace(10) delece(3) (jednou současně přestavba MLL) 4
61 Tabulka 17. Porovnání klinických a cytogenetických dat u nemocných dle identifikovaného rekurentního zlomu na chromosomu 11. Oblast zlomu Lokalializace (Mb) Geny Frekvence* Ø věk Poměr Ž/M Poměr P/S FAB typ (počet) 11p15.4 ch11: NUP98 3/5 % 58 1/2 3/0 M2(2) M4(1) 11p13 ch11: DCDC5, DCDC1 4/7 % 64 1/3 4/0 M4(2) M0(1) M2(1) 11p12 ch11: žádné 2/4 % 71 1/1 2/0 M0(1) M1(1) 11q13.2 ch11: GAL, MTL5, 2/4 % 54 0/2 2/0 M2(1) lincrna M5(1) 11q23.3 ch11: MLL 28/52 % 46 15/13 20/8 M5(14) M1(7) M4(4) M2(2) Mechanismus zlomu (počet) Komplexní karyotyp Dosažení 1. remise alo- TKD žije Ø doba přežití (měsíce) BP(2) duplikace(1) BP(1) NP(2) delece (1) BP(1) delece(1) BP(2) jiné(1) Vysvětlivky: * % z počtu nemocných s abnormalitou chromosomu 11; Ž, ženy; M, muži, P, primární; S, sekundární; BP, balancovaná přestavba; NP, nebalancovaná přestavba; alo-tkd, alogenní transplantace kostní dřeně. BP(25) NP(3)
62 5.2. Změny chromosomu 11 u dětí s AML Změnu chromosomu 11 jsme prokázali u pěti (36 %) ze 14 dětí s nově diagnostikovanou AML (klinická data viz tabulka 18). U všech byla klasickou cytogenetickou analýzou prokázána reciproká přestavba oblasti 11q23.3. Metoda FISH potvrdila přestavbu genu MLL u všech nemocných. Klinická a cytogenetická data nemocných jsou zobrazeny v tabulce 18. Tabulka 18. Přehled klinických a cytogenetických dat u dětských pacientů s AML. Pacient Pohl/věk Datum dg. AML Karyotyp Zlom alotkd Přežití (dny)* 1 Ž/ M5 46,XX,t(11;19)(q23;p13.3)[18] MLL ANO 978¹ 2 Ž/ M5 46,XX,t(3;9;11)(p21;p22;q23.3)[22] MLL NE 376¹ 3 M/ M5 46,XY,t(9;11)(p22;q23)[12] MLL NE 98¹ 4 M/ M5 46,XY,t(9;11)(p22;q23)[2] MLL NE 64¹ 5 Ž/ M5 46,XX,t(9;11)(p22;q23)[22] MLL NE 43¹ Vysvětlivky: *, k datu ; ¹, žijící Skríningové vyšetření u AML Základem molekulárně cytogenetického skríningového vyšetření byla detekce přestaveb MLL genu, která byla provedena u všech 300 dospělých nemocných. Dále jsme se u 245 nemocných zaměřili na přítomnost delecí dlouhých ramen chromosomů 5 a 7 s využitím lokus specifických sond LSI 5q31/5p15.2 a LSI 7q31/CEP 7, a na detekci trisomie chromosomu 8 pomocí centromerické sondy CEP 8. K ní byla jako kontrolní obvykle použita centromerická sonda pro chromosom 9. Změnu MLL genu jsme prokázali u 45 nemocných (15 %). Ve 25 případech se jednalo o balancovanou přestavbu, u tří nemocných o nebalancovanou, podrobnosti byly popsány v kapitole Nadpočetné kopie celého MLL genu, tedy více než dva fúzní signály detekční sondy, jsme zjistili u 17 pacientů. Tři kopie MLL genu v rámci parciální nebo úplné trisomie 11 jsme prokázali u jedenácti nemocných, amplifikaci MLL genu (čtyři a více kopií) se nacházela u šesti pacientů, u všech současně s mnohočetnými změnami dalších chromosomů. Deleci chromosomu 5 oblasti q31 jsme prokázali u 30 pacientů (12 %). Ve dvou případech se del(5)(q31) nepodařilo prokázat klasickou cytogenetickou analýzou, pouze metodou FISH. Deleci oblasti 7q31 nebo monosomii chromosomu 7 jsme detekovali u 28 pacientů (11 %). U čtyř nemocných byla změna chromosomu 7 odhalena jen metodou
63 FISH (dvakrát se jednalo o nález 0 mitos, jednou byla klasickou cytogenetickou analýzou prokázána pouze samostatná del(5)(q31) a jednou samostatná trisomie 8). Trisomii chromosomu 8 jsme prokázali u 24 nemocných (10 %). Dvakrát byla trisomie 8 prokázána pouze metodou FISH, jednou se jednalo o nepřítomnost dělících se buněk (nález 0 mitos), u druhého byl nález z klasické cytogenetické analýzy normální. U 238 nemocných byla jako kontrolní sonda pro detekci trisomie chromosomu 8 použita centromerická sonda pro chromosom 9. Početní odchylky chromosomu 9 jsme v rámci skríningu prokázali u pěti nemocných (4 %). Ve čtyřech případech se jednalo o nadpočetnou kopii chromosomu 9 a jednou o monosomii 9. Graf 7. Výsledky FISH skríningového vyšetření u dospělých nemocných s AML přestavba MLL nadpočetná kopie MLL del(5)(q31) del(7)(q31)/-7 trisomie 8 trisomie/monosomie 9 Vysvětlivky: osa x (abnormalita), osa y (počet nemocných), modřě patologický nález. 63
64 6. DISKUZE Změny chromosomu 11 byly popsány u řady hematologických onemocnění. Často se vyskytují například u akutní myeloidní leukémie. Pro naší studii jsme proto zvolili soubor nemocných právě s touto diagnózou. AML je vemi heterogenní onemocnění, a to z hlediska fenotypového i genetického. Průměrný věk nemocných s AML se pohybuje mezi 63 a 65 lety (Mayer a Starý, 2002). V našem souboru dospělých nemocných se často vyskytovali mladší pacienti, průměrný věk v době stanovení diagnózy byl 55 let. Důvodem tohoto rozporu je výběr nemocných do léčebného programu ÚHKT, do kterého bývají přijati především nemocní mladšího věku, u kterých je provedení transplantace kostní dřeně méně rizikové. Mezi časté AML podtypy dle FAB klasifikace patří M2, M4 a M1. Také v našem souboru se tyto FAB typy vyskytovaly nejčastěji: M2 u 28 %, M1 u 20 % a M4 u 19 % nemocných. Při diagnostice onemocnění se v současné době klade zvláštní důraz na cytogenetický nález, protože mnoho chromosomových aberací je spojeno i s určitým prognostickým významem (Bloomfield a kol. 1998, Mrózek a kol. 2001). Uvádí se, že přes 50 % pacientů s AML má při diagnóze abnormální karyotyp (Mrózek a kol. 2001, Schoch a kol. 2003, Marcucci a kol. 2004). V našem souboru dospělých nemocných jsme na základě kombinace klasické a molekulárně - cytogenetické analýzy prokázali chromosomové změny u 51 % pacientů, normální karyotyp u 36 % a ve 13 % případů nebyly nalezeny žádné dělící se buňky nebo bylo přítomno méně než 10 normálních mitos. Ve shodě s literaturou jsme jako nejčastější strukturní aberace detekovali translokaci 11q23.3, del(7)(q?), t(8;21)(q22;q22), inv(16)(p13q22)/t(16;16)(p13;q22), del(5)(q?) a t(3;3)(q21;q26)/ins(3;3)(q26;q21q26). Početní odchylky nejvíce postihovaly chromosomy 7, 8, 13 a 21. Komplexní přestavby karyotypu jsou detekovány u 10 až 15 % nemocných s AML (Mrózek a kol. 2001, Babická a kol. 2007). V naší studii jsme nález prokázali u 17 % nemocných. Dle Babické a kol. (2007) vstupují nejčastěji do komplexních přestaveb karyotypu chromosomy 5, 7 a 11. Podobnou asociaci prokázali Limberg a kol. (2002) a můžeme ji potvrdit i v našem souboru. Oproti dospělým nemocným s AML je u dětských pacientů v literatuře udáván patologický karyotyp až v 80 % případů (Mrózek a kol. 2004, Manola 2009). Také v našem souboru nemocných dětí jsme získané chromosomové aberace detekovali u 86 %, nejčastěji se jednalo o přestavbu genu MLL (n=5). 64
65 Prokázali jsme, že změny chromosomu 11 patří mezi časté cytogenetické nálezy u dospělých nemocných s nově diagnostikovanou AML s frekvencí výskytu 18 %. Aberace postihovaly častěji dlouhé rameno chromosomu než krátké. Nestabilitu chromosomu 11 u AML jsme potvrdili nálezem dvou a více zlomů přítomných na stejném chromosomu 11 u jedenácti nemocných. Ve dvou případech byly postižny oba homologní chromosomy 11. Naopak u pěti nemocných byla prokázána trisomie chromosomu 11, tedy žádné zlomové místo. Pomocí kombinace klasické cytogenetické analýzy, molekulárně cytogenetické analýzy a SNP array jsme na derivovaných chromosomech 11 odhalili více než 35 různých zlomů. K zlomům došlo jak na základě balancovaných, tak i nebalancovaných změn. Alteraci krátkého ramene chromosomu 11 jsme prokázali u 24 % nemocných se změnou chromosomu 11. Z klinického hlediska tvořili nemocní poměrně homogenní skupinu starších pacientů s průměrným věkem při diagnóze nad 60 let. AML FAB subtyp odpovídal převážně typu M2 a M4, oba patří mezi obecně nejčastější FAB subtypy u AML. Sekundární forma onemocnění, která byla diagnostikována u 31 % nemocných s abnormalitou 11p, byla jen o málo vyšší než incidence v celkovém souboru (24 %). Pro nemocné se změnou 11p byla charakteristická velmi krátká doba přežití, v průměru čtyři měsíce. Většině nemocných se nepodařilo dosáhnout ani první kompletní remise. Pokud k tomu došlo, následoval brzy relaps onemocnění. Velmi nepříznivá prognóza mohla být ovlivněna i dalšími chromosomovými změnami a jejich formou. Z cytogenetické hlediska byl typickým nálezem nemocných komplexní karyotyp a nebalancovaný charakter změn 11p, a to zejména vlivem delece. Na krátkém rameni chromosomu 11 jsme neprokázali žádnou společně deletovanou oblast, která by chyběla u všech nemocných, přesto byly některé postiženy častěji: 11pter-11p15.5 (n=4; ch11: Mb), 11p p13 (n=4; ch11: Mb) a 11p13 (n=4; ch11: Mb). V telomerické a subtelomerické oblasti 11p je lokalizována řada známých a předpokládaných tumor supresorových genů jako je například MIR210 (mikrorna inhibující přežití a proliferaci nádorových buněk), MUC6, MUC2, MUC5B (muciny uplatňující se při proliferaci a vzniku metastáz) a CDKN1C (inhibitor cyclin dependentní kinázy, p57). V oblasti se vyskytuje i řada genů, jejichž proteiny se uplatňují v transdukci signálu (HRAS, CD151) nebo jsou součástí ribozomu (RPLP2). Důležitou úlohu ztráty ribozomového proteinu jako kritického bodu při vzniku tvz. 5q- syndromu (myelodysplastický syndrom s izolovanou del(5)(q) a s charakteristickými klinickými projevy) popsali Boultwood a 65
66 kol. (2010) a Jadersten a kol. (2010). Nejen delece ale i ztráty heterozygozity oblasti 11p15 jsou popisovány u mnoha různých nádorů. Geny lokalizované v této oblasti jsou proto často cílem studií pro vývin protinádorové léčby (Gupta a kol. 2008, Jordheim a kol. 2011). Naopak v deletované oblasti 11p14.1-p13 a 11p13 se vyskytuje jen několik málo genů například MPPED2 (inhibitor proliferace), DCDC5, DCDC1 (úloha při polymerizaci mikrotubulinu), DNAJC24 (stimulátor ATPáz) nebo IMMPL (mitochondriální peptidáza). U žádného z těchto genů zatím nebyla prokázána asociace se vznikem malignity. Bez ohledu na mechanismus vzniku zlomového místa jsme pomocí FISH mapování na 11p identifikovali 18 různých zlomových míst. Rekurentní zlom byl prokázán v oblasti 11p15.4 v genu NUP98. Další potencionálně nenáhodné zlomy jsme specifikovali do oblastí: 11p13 a 11p12. Přestavbu genu NUP98 (11p15.4) jsme prokázali u tří nemocných. U všech byly součástí komplexních změn karyotypu. Ve dvou případech se jednalo o variantní komplexní translokace t(5;11;20)(q35;p15;p?) a t(4;5;11)(q11;q35;p15). Translokace t(5;11)(q35;p15) byla jako rekurentní chromosomová abnormalita poprvé popsána u dětí s de novo AML se zdánlivě normálním karyotypem nebo delecí del(5q) jako samostatnou aberací (Jaju a kol. 2001). Později ji další autoři prokázali i u dospělých nemocných s AML (Casas a kol. 2003). Jako partnerský gen na chromosomu 5 byl identifikován gen NSD1 (5q35). Také v obou našich případech jsme prokázali fúzi obou genů. Předpokládá se, že výsledný chimérický protein je tvořen interakční doménou jaderného receptoru NSD1 a RNA vazebnými doménami NUP98. Protože se oba geny nachází v blízkosti telomer, je translokace klasickou cytogenetickou analýzou nedetekovatelná, a to i v rámci komplexního karyotypu - jak dokládáme. Často ji nelze prokázat ani metodami mfish nebo mband. Jistotu detekce poskytuje pouze metoda FISH. Kryptickou komplexní translokaci t(3;5;11)(p25;q35;p15), původně definovanou jako jednoduchá reciproká translokace t(3;5), odhalili precizním mapováním zlomových míst i Ishikawa a kol. (2007). Další komplexní translokaci t(10;20;11)(q24;q11;p15) popsal Panagopoulos a kol. (2002). Jako fúzní partner genu NUP98 byl identifikován gen TOP1 (20q11). Protože u nemocného č. 10 byl chromosom 20 též postižen, otestovali jsme alteraci tohoto genu metodou FISH s BAC sondou. Přestavba genu TOP1 ale nebyla potvrzena. U třetí nemocné byla přestavba genu NUP98 nebalancovaná. Vzhledem k vzácnému výskytu je stanovení prognózy 66
67 NUP98 přestaveb velmi obtížné. Recentní práce zatím stále více poukazují na spojitost přestaveb NUP98 s nepříznivou prognózou pro nemocné, krátkou dobou přežití a špatnou odpovědí na léčbu, jak u dospělých tak i dětských pacientů (Hollink a kol. 2011, Lundin a kol. 2011). Hollink a kol. (2011) ve své studii zabývající se charakteristikou NUP98-NSD1 fúzního genu prokázal, že nemocní tvoří homogenní skupinu mladších pacientů specifickou nepříznivým klinickým průběhem. Doporučil proto detekci této abnormality v rámci rutiního skríningu. U našich dvou nemocných byla průměrná doba přežití vyšší než u ostatních nemocných s abnormalitou 11p. U nemocného č. 10 byl současně postižen i druhý homologní chromosom 11 a u nemocného č. 54 se na stejném chromosomu 11 nalézal další zlom. Je tak otázkou nakolik byl stav nemocných ovlivněn také přítomností dalších zlomů. V oblasti 11p13 jsme identifikovali tři překrývající se místa, do nichž byl specifikován zlom: ch11: Mb, ch11: Mb a ch11: Mb. První region zahrnuje pouze geny DCDC5 a DCDC1. Ve druhém a třetím regionu se sice nachází i několik dalších genů, ale součástí oblasti jsou opět i geny DCDC5 a DCDC1. Oba geny mají významnou roli při polymerizaci mikrotubulů, která je nezbytná pro dělení buňky a cytoplasmatický transport. Geny jsme proto vyhodnotili jako důležité kandidáty. V potencionálně nenáhodném zlomu v chromosomovém pruhu 11p12 (ch11: Mb) nebyl zatím prokázán ani není předpokládán žádný protein nebo RNA kódující gen. Role tohoto zlomového místa tak zůstavá velmi diskutabilní. Může se jednat o repetitivní sekvence, u kterých dochází obecně často k skluzu polymerázy avšak bez poškození konkrétního genu anebo sekvence regulující transkripci genů lokalizovaných mimo region. Další překrývající se místa na 11p, do nichž byl specifikován zlom, jsme identifikovali také v regionech: 11p15.5 (ch11: Mb), 11p15.4-p15.1 (ch11: Mb) a 11p12 ( Mb). Z důvodu nedostatku dělících se buněk kostní dřeně však tyto oblasti nebyly zcela specifikovány a zůstaly příliš velké na to, abychom je mohli s určitostí označit za rekurentní. Vzhledem k malému překryvu místa, do něhož byly zlomy specifikovány v oblasti 11p12, předpokládáme i v tomto případě spíše dva různé zlomy. V jednom případě jsme prokázali zlom v genu LRRC4 (11p12). Protože v literatuře dosud nebyl tento gen zmíněn v souvislosti s hematologickou malignitou, 67
68 předpokládáme, že se jedná o náhodný zlom bez významné role v leukemogenezi. Hypotézu je ale nezbytné ověřit na větším souboru nemocných. Změnu dlouhého ramene chromosomu 11 jsme prokázali u 40 nemocných. Literatura doporučuje odlišovat přestavby MLL genu jako samostatný specifický soubor od ostatních změn 11q (Döhner a kol. 2010, Marchesi a kol. 2011). Proto jsme i v naší studii abnormality oddělili a hodnotili zvlášť. Alteraci 11q bez účasti genu MLL jsme podobně jako abnormality 11p identifikovali u 26 % nemocných. I tito nemocní tvořili z klinického hlediska poměrně homogenní skupinu, a to starších pacientů s průměrným věkem při diagnóze nad 60 let. AML FAB subtyp opět odpovídal převážně typu M2, tedy obecně nejčastějšímu FAB subtypu u AML. Sekundární forma onemocnění byla diagnostikována u 29 % nemocných s abnormalitou 11q nezahrnující přestavbu MLL genu. Přestože se u šesti nemocných podařilo dosáhnout první kompletní remise, doba přežití nemocných se změnou 11q byla velmi krátká, stejně jako u 11p změn činila v průměru čtyři měsíce. Také v případě 11q alterací se na nepříznivé prognóze podílely i další chromosomové změny a nebalancovaná forma postižení 11q. U 71 % nemocných byl nález součástí komplexního karyotypu. Jednoznačně převažující nebalancovanou formou alterací 11q byly amplifikace a duplikace. Při amplifikaci dochází k vzniku mnohonásobných kopií genu a následné nadprodukci onkoproteinu. Předpokládá se, že amplifikace onkogenů mají důležitou roli v nádorové progresi a bývají typickým znakem metastazujících buněk (Hesketh 1997). U hematologických malignit se dlouho jevily jako vzácný jev. Tanaka a kol. (1993) detekovali genové amplifikace jen u přibližně 1 % případů AML. Pozdější i naše studie však poukazují na vyšší procento nálezu této aberace při využití metod molekulární cytogenetiky (Michaux a kol. 2000, Papenhausen a kol. 2005, Sarova a kol. 2010). Pouze u jednoho nemocného postihovala amplifikace oblast krátkých ramen a nikoliv současně i dlouhých. Společnou oblastí duplikace byla 11q q24 (n=10; ch11: Mb), začínající od 3 konce genu MLL. Společnou oblastí amplifikace na 11q (více než tři kopie) byl u všech nemocných 5 konec MLL genu. Dále byl často amplifikován úsek 11q q25. Užitím acgh, Rucker a kol. (2006) identifikovali tři společně amplifikované regiony na 11q: 11q12-q14, 11q23.3 a 11q23.3-q24.1. Zatkova a kol. (2009) detekovali devět rekurentních amplifikovaných oblastí, tři z nich byly zmnoženy u všech nemocných: 11q23.3, 11q24.2-q24.3 a 68
69 11q24.3-q25. Lze tedy shrnout, že součástí amplikonu je skoro vždy gen MLL nebo jeho části a distálně orientované oblasti. V často zmnožené oblasti 11q q25 je lokalizována řada důležitých genů. Kromě proto-onkogenu MLL se zde nachází i další geny uplatňující se při transdukci signálu NFRKB, ARHGEF12 nebo SPA17. V patogenezi AML byl již gen ARHGEF12 popsán při fúzi s MLL genem vlivem intersticiální delece 11q23 (Kourlas a kol. 2000). Specifickou funkci s možnou úlohou při leukemogenezi mají také geny DDX6 (RNA helikáza), H2AFX (histon), ETS1 (inhibitor apoptózy), UBE4A (ubiquitinylace) nebo mir-100. Gen mir-100 kóduje mikrorna (mirna). Ta je definována jako malá nekódující RNA o velikosti přibližně 22 nukleotidů. mirna hrají velmi důležitou roli při diferenciaci buněk a při iniciaci a progresi nádoru. Wooi Loon Ng a kol. (2010) sledoval inhibiční účinek zvýšené exprese mir-100 na expresi tumor supresorového genu ATM, lokalizovaného také na chromosomu 11 (11q22). Serin-treoninová kináza ATM je nezbytná při procesech homologní i nehomologní rekombinace. Účast amplifikace MLL genu u AML potvrdilo i mnoho dalších studií (Poppe a kol. 2004, Rucker a kol. 2006, Zatkova a kol. 2009). U nemocných s amplifikací nebo nadpočetnou kopií MLL genu popisují Schnittger a kol. (2000) a Arnaud a kol. (2005) dominanci FAB subtypu M2. Také v našem souboru jsme zaznamenali duplikaci/amplifikaci MLL genu nejčastěji u tohoto subtypu. Amplifikace genu je u myeloidních onemocnění často asociována se sekundárním typem onemocnění, s komplexními změnami karyotypu, zahrnujícími obvykle del(5q) a agresivním typem onemocnění s velmi špatnou odezvou na léčbu a krátkou dobou přežití. (Andersen a kol. 2001, Březinová a kol. 2002, Papenhauesen a kol. 2005, Herry a kol. 2006). Také v našem souboru jsme u všech šesti pacientů s více než třemi kopiemi genu současně prokázali rozsáhlé změny karyotypu, pouze u tří nebyla přítomna delece del(5q). Velmi špatný klinický průběh nemocných s amplifikací MLL genu oproti jeho duplikacím a balancovaným změnám jsme prokázali i v našem souboru. Rucker a kol. (2012) uvádí, že amplifikace oblastí chromosomu 11 je také často specificky provázena molekulárními mutacemi genu TP53. Mutace tohoto tumor supresorového genu patří mezi obecně známé nepříznivé faktory a může tak umocňovat i velmi špatný klinický průběh u nemocných s amplifikací chromosomu 11. Aventín a kol. (2003) popsal dva případy parciální trisomie chromosomu 11 s nadpočetnou kopií MLL genu, která byla zároveň detekována spolu s jeho interní duplikací. Podobně Pajuelo-Gámez a kol. (2007) pozoroval případy komplexních změn 69
70 karyotypu s mnohonásobnou amplifikací MLL genu dle FISH, u nichž byla pomocí RT- PCR zjištěna současně parciální tandemová duplikace MLL. V našem souboru jsme MLL PTD detekovali spolu s parciální trisomií chromosomu 11 pouze u jednoho nemocného (nemocný č. 45). Různorodost formy duplikací/amplifikací chromosomu 11 jsme také publikovali (Sarova a kol. 2010). Bez ohledu na mechanizmus vzniku zlomového místa jsme pomocí FISH mapování na 11q identifikovali 21 různých zlomových míst. Potencionálně nenáhodný zlom jsme specifikovali do oblasti 11q13.2. V místě zlomu 11q13.2 se nachází dva kandidátní geny: GAL a MTL5. Galanin je neuropetid, který se uplatňuje při transferu molekul mezi centrálním a periferním nervovým systémem. Jeho deregulace byla již popsána u některých solidních nádorů (Rauch and Kofler 2010). Gen MTL5 se uplatňuje při spermatogenezi, ale také při buněčném růstu a diferenciaci. U obou nemocných s alterací 11q13.2 byl současně přítomen i další zlom na chromosomu 11 v oblasti 11p15.4 (v genu NUP98) a v oblasti 11q23.3 (v genu MLL). Stanovení prognostického významu je tedy pro tento zlom nejasné. Překrývající se oblasti, do nichž byl zlom specifikován, jsme identifikovali také v regionech: 11q12 (nemapováno), 11q14.2-q21 ( Mb) a 11q24 (nemapováno). I v těchto případech se však z důvodu nedostatku dělících se buněk kostní dřeně nepodařilo zlomová místa zcela specifikovat a zůstala tak příliš velká na to, abychom je mohli s určitostí označit za rekurentní. Ve dvou případech pokrývavalo místo zlomu pouze jeden protein-kódující gen: ODZ4 (11q14.1) a MAML2 (11q21). Alterace ODZ4 genu byla prokázána pouze jednou a také v literatuře dosud nebyla popsána v souvislosti s hematologickou malignitou. Předpokládáme proto, že se jedná o náhodný zlom. Dvě překrývající se místa, do níž byl specifikován zlom, byla pozorována v oblasti: 11q21: ch11: Mb zahrnující pouze gen MAML2 a ch11: Mb pokrývající kromě MAML2 genu přibližně 30 dalších genů. Jako fúzní partner genu MLL byl MAML2 již popsán u nemocných s AML a MDS (Nemoto a kol. 2007). Protein genu slouží jako transkripční ko-aktivátor pro NOTCH receptory, čímž se podílí na transdukci signálu. Vzhledem k příliš velké oblasti, do níž bylo specifikováno druhé zlomové místo, je i v tomto případě vyhodnocení alterace genu MAML2 jako rekurentní v našem souboru nemožné. Lze jej tak považovat jen na základě publikovaných dat. 70
71 Mnoho míst, do nichž byl specifikován zlom na 11p i 11q, nepokrývaly pouze protein kódující geny, ale často také některé mikrorna (mirna) nebo dlouhé nekódující RNA (lncrna). Regulační role mirna především jako supresorů genové exprese byla již prokázána a je předpokládána u více než 60 % protein kódujících genů. Oproti tomu lncrna zatím nejsou tak dobře prostudovány (Nikitina a kol. 2012). Podobně jako uvádí literatura (Braekeleer a kol. 2005, Meyer a kol. 2009), byl i v našem souboru nemocných identifikován gen MLL (11q23.3) jako nejčastější zlomové místo na chromosomu 11 vůbec, a to u poloviny ze všech nemocných se změnou chromosomu 11. Aberace MLL genu jsou popisovány u 5 až 10 % pacientů s de novo AML a 25 % pacientů se sekundárním typem onemocnění (Rowley a kol. 1999). V našem souboru jsme přestavbu MLL genu detekovali pouze u 11 % nemocných se sekundární formou AML. Průměrný věk pacientů v době stanovení diagnózy byl 46 let, což je přibližně o 15 let méně než u nemocných s ostatními změnani chromosomu 11. Také Schoch a kol. (2003) ve své studii zahrnující 54 nemocných s přestavbou MLL genu uvádí značně vyšší incidenci u pacientů mladších 60 let. Většina publikovaných případů AML s přestavbou MLL genu odpovídala dle FAB kritéria AML M4 nebo AML M5a (Ayton a Cleary 2001, Schoch a kol. 2003). Poirel a kol. (1996) udává spojitost MLL přestaveb i se subtypem M1. V našem souboru byly nemocní nejčastěji diagnostikováni jako AML M5a, M1 a M4. Většina translokací MLL genu je balancovaná a dává vznik dvěma leukemogenním chimérickým genům. Při pokusech na myších modelech byl prokázán jako kritický fúzní gen tvořený 5 koncem MLL genu a 3 koncem partnerského genu. Obvykle bývá lokalizován na derivovaném chromosomu 11 (Ayton a Cleary 2001, Daser a Rabbitts 2004). Vzhledem k tomu, že se přestavby MLL genu vyskytují u hematologických malignit myeloidní i lymfatické řady buněk, předpokládá se, že se partnerské fúzní geny uplatňují v určení fenotypu leukémie (Collins a Rabbitts 2002). V našem souboru jsme se nejčastěji setkali s translokacemi t(9;11)(p22;q23) a t(11;19)(p13;q23). Reciprokou translokaci t(9;11)(p22;q23) jsme prokázali u 11 nemocných. Jedná se o nejčastěji popisovanou translokaci MLL genu u AML, při které dochází k fúzi 5 konce MLL genu s 3 koncem genu MLLT3 (AF9) (Schoch a kol. 2003, Braekeleer a kol. 2005). Uvádí se, že v 70 % případů je přestavba nalézána jako samostatná aberace, u 20 % případů se vyskytuje spolu s trisomií chromosomu 8 (Mrózek a kol. 1997). Tuto spojitost jsme potvrdili u tří nemocných. 71
72 Schoch a kol. (2003) detekovali přídatné chromosomové aberace u 41 % nemocných s přestavbou MLL genu. Prokázali také, že tyto přídatné změny nemají vliv na prognózu, která je vždy špatná. Meyer a kol. (2009) ve své studii rozděluje mnohočetné přestavby zahrnující translokaci MLL genu do dvou skupin. První zahrnuje inverze a inzerce nutné k vzniku funkčního chimérického proteinu při neshodném směru transkripce fúzních genů vzhledem k jejich orientaci na chromosomu, druhou tvoří komplexní translokace. Příkladem fúze transkripčně opačně orientovaných genů je translokace t(10;11)(p12;q23), vedoucí k MLL-MLLT10 (AF10) fúznímu genu. Zatímco gen MLL je transkripčně aktivní ve směru od centromery k telomeře, partnerský gen MLLT10 je přepisován ve směru opačném. Jednoduchá fúze by tedy vedla na chromosomu 11 k vzniku fúzního genu složeného z 5 částí obou genů. Vznik funkčního chimérického transkriptu proto vyžaduje nejméně tři chromosomové zlomy. Fúze obou genů jsou tedy často doprovázeny dalšími aberacemi (Limbergen a kol. 2002, Matsuda a kol. 2006). Celkem bylo popsáno 6 různých mechanizmů vedoucích k MLL-MLLT10 fúznímu transkriptu. Translokaci může předcházet paracentrická inverze části 11q, obvykle 11q13/21-23, nebo naopak (mikro)inverze části MLLT10 genu, přesunuté na derivovaný chromosom 11. Pozorovány byly také inzerce asi megabázového regionu zahrnujícího 5 konec MLL genu do krátkých ramen chromosomu 10, případně 10p do 11q anebo kryptické přestavby v MLL genu (Klaus a kol. 2003). Inzerci 5 konce MLL genu do 10p12 jsme prokázali i v našem souboru nemocných, a to u pacienta č. 26. Inzerce nebyla detekovatelná metodou mfish. Kryptické inzerce 5 konce MLL genu byly popsány i do dalších chromosomů například do 6q, 9p a dalších (Dyson a kol. 2003, Douet-Guilbert kol. 2005). Dalším mechanizmem je inzerce amplifikovaného 5 konce MLL genu bez jakéhokoliv přeskupení či ztráty obou kopií MLL genu na obou homologních chromosomech 11 (Jarošová a kol. 2005). Tento způsob předpokládáme i u naší pacientky č.18 s přítomností dvou patologických klonů s amplifikavaným 5 koncem MLL genu inzertovaným do oblasti 10p12 a dvěmi normálními kopiemi MLL genu. Podobné přestavby s opačně orientovanými geny byly pozorovány i u solidních nádorů nebo i dalších typů leukémií jako je například chronická myeloidní leukémie a fúze genů ETV6 (12p13) a ABL1 (9q34) (Klaus a kol. 2003). Druhá skupina komplexních přestaveb zahrnující MLL gen je tvořena reciprokými translokacemi mezi třemi chromosomy, při nichž dochází k fúzi 5 konce MLL genu s jedním z mnoha frekventovaných partnerských genů a fúzi 3 konce genu s novým 72
73 genem nebo je 3 konec MLL genu vložen do chromosomové oblasti nekódující žádný gen. Dle databáze Atlas of Genetics and Cytogenetics in Oncology and Hematology do komplexních translokací ponejvíce vstupuje kromě chromosomu 11 také chromosom 9 ( k datu ). Třetí chromosom bývá variabilní. V našem souboru jsme komplexní přestavbu zahrnující tři chromosomy identifikovali u dvou nemocných č. 17 a 19. Jednalo se o translokace t(7;11;10)(q21;q23;p12) a t(9;12;11)(p22;p13;q23), tedy variantní formy frekventovaných translokací t(10;11)(p12;q23.3) a t(9;11)(p22;q23.3), k nimž přibyly partnerské chromosomy 7 a 12. Chromosomový pruh 7q22 patří mezi často deletované regiony na chromosomu 7 u hematologických malignit, předpokládá se, že je zde lokalizována řada nádorových supresorových genů, jejichž inaktivace má význam v patogenezi myeloidních onemocnění (Curtiss a kol. 2005, Březinová a kol. 2007). Na chromosomu 12 jsme v oblasti 12p13 prokázali zlom v genu ETV6. Gen ETV6 je důležitým transkripčním regulátorem s významnou rolí v hematopoese. Podobně jako gen MLL má i on mnoho partnerských genů. Jeho mutace se vyskytují nejen u leukémií a myelodysplastických onemocnění, ale i u sarkomů (Viera a kol. 2005). Variantní formy častých jednoduchých reciprokých translokací byly popsány i v mnoha dalších studiích (Vieira a kol. 2006, De Braekeleer a kol. 2010). Ve sledovaném souboru nemocných s alterací MLL genu jsme také identifikovali novou dosud nepublikovanou translokaci t(6;11)(p21.3;q23). Na chromosomu 6 byly popsány čtyři partnerské geny, a to vždy na jeho dlouhém rameni. V našem případě jsme konkrétní partnerský gen neodhalili, jen jeho lokalizaci specifikovali do oblasti 6p21.3, centromericky k regionu hybridizujícímu k BAC sondě RP11-174A4. Zajímavostí je, že translokace se vyskytovala v rámci tetraploidního karyotypu. Hyperploidie a tetraploidie patří mezi vzácné chromosomové změny u AML a MDS (Iyer a kol. 2004). Nacházeny bývají u starších mužů s de novo typem onemocnění. Charakterizovány jsou krátkou dobou přežití a častým nedosažením remise, čímž se řadí do skupiny prognosticky velmi nepříznivých cytogenetických abnormalit (Iyer a kol. 2004). V našem případě se jednalo o ženu středního věku s doposud dobrým klinickým průběhem (přežití k činilo 246 dní). Z počátku byly přestavby genu MLL řazeny do středně příznivé prognostické skupiny (Grimwade a kol. 1998). Mnoho pozdějších studií ale poukázali na spojitost s prognózou nepříznivou. Výjimku tvoří pouze translokace t(9;11)(p22;q23), u které byl prokázán o něco lepší klinický průběh onemocnění než u ostatních translokací 73
74 (Grimwade a kol. 2010, Marchesi a kol. 2010). V našem souboru nemocných činila průměrná doba přežití 15 měsíců. Neprokázali jsme statisticky významný rozdíl mezi přežitím nemocných s různými MLL translokacemi. To může být zapříčiněno tím, že studie, které rozdíl zaznamenaly, byly založeny na větších souborech nemocných. Odlišné přežití jsme nepozorovali ani při porovnání skupiny nemocných s normálním karyotypem a nemocných s přestavbou MLL genu, což lze vysvětlit častou indikací pacientů s přestavbou MLL genu k provedení transplantace kostní dřeně. Ta byla v našem souboru provedena skoro u poloviny nemocných s přestavbou genu MLL. Také ve skupině nemocných s normálním karyotypem je možná přítomnost některé nepříznivé molekulární mutace, která je cytogeneticky nedetekovatelná, ale může zhoršit prognózu těchto nemocných. Statisticky významný rozdíl se nám nepodařilo prokázat ani při porovnání balancované a nebalancované přestavby MLL genu nebo přestavbou MLL genu jako samostatné abnormality či s jednou početní odchylkou v porovnání s případy s mnohočetnými přídatnými chromosomovými změnami. Tento výsledek je v souladu s literaturou, která uvádí, že přídatné chromosomové aberace neovlivňují prognostický význam MLL přestaveb (Marchesi a kol. 2010). Naopak významný rozdíl v přežití jsme sledovali u nemocných s různým typem MLL alterace. Nemocní s nadpočetnou kopií MLL genu měli kratší dobu přežití než nemocní s jeho balancovanou přestavbou. V našem souboru nemocných se při dodržení stávající léčebné strategie ukázaly přestavby MLL genu jako ukazatel lepšího klinického průběhu v porovnání s ostatními změnami chromosomu 11. Potvrdili jsme, že přestavby genu MLL tvoří velmi specifický soubor nemocných s podobným klinickým stavem i charakterem onemocnění. Proto se tento gen stává často těžištěm analýz pro vytvoření nové cílené terapie. Velké množství heterogenních partnerských genů však znesnadňuje a velmi komplikuje snahu vědeckých pracovišť o vytvoření universální terapie pro MLL iniciované leukémie. Mnoho z nich je totiž nově popsaných a není u nich zatím známa jejich funkce. Je proto třeba ještě mnoha dalších molekulárních studií k porozumění funkce MLL genu a jeho fúzních partnerských genů v patogenezi akutních leukémií. Alterace MLL genu jsme vyšetřovali i na molekulární úrovni v podobě parciální tandemové duplikace (PTD). MLL PTD je popisována u přibližně 5 až 9 % případů AML (Dohner a kol. 2002, Mrózek a kol. 2007). V našem souboru nemocných jsme MLL PTD prokázali u 23 % nemocných s AML. Tento nesoulad může být způsoben zaměřením řady studií pouze na nemocné s normálním karyotypem, u nichž je MLL 74
75 PTD nejčastější. Také v našem souboru tvořila největší část nemocných s MLL PTD skupina s normálním karyotypem. Druhým nejfrekventovanějším cytogenetickým nálezem spojeným s výskytem s MLL PTD je trisomie chromosomu 11 (Caligiuri a kol. 1996). Tuto souvislost můžeme potvrdit přítomností MLL PTD u dvou z pěti případů s trisomií chromosomu 11. Schnitger a kol. (2000) ve své studii nenašli žádnou spojitost s konkrétním FAB subtypem AML. Také my jsme MLL PTD sledovali převážně u obecně nejčastěji se vyskytujících FAB subtypů M2, M1 a M4. Schnitger a kol. (2000) nedetekovali MLL PTD u žádného pacienta s prognosticky příznovou cytogenetickou změnou, rekurentní translokací nebo komplexním karyotypem. Naproti tomu v našem souboru jsme MLL PTD prokázali u čtyř nemocných s prognosticky příznivou aberací, u pěti nemocných s komplexním karyotypem a u sedmi pacientů s přestavbou genu MLL, především s translokací t(9;11)(p22;q23). Při statistické analýze jsme však neprokázali, že by přítomnost MLL PTD měla vliv na přežití nemocných s přestavbou MLL. Nejčastěji dochází k duplikaci exonů 5 až 11 (12) nebo exonů 2 až 6, což jsme potvrdili i v naší studii (Whitman a kol. 2005). Literatura uvádí nepříznivý prognostický význam u MLL PTD (Grimwade a Hills 2009). V našem souboru jsme odlišnost mezi přežítím nemocných s normálním karyotypem a MLL PTD- a normálním karyotypem a MLL PTD+ neprokázali. Důvodem může být opět častá indikace pacientů s MLL PTD k provedení transplantace kostní dřeně a také možná přítomnost nepříznivé molekulární mutace ve skupině nemocných s normálním karyotypem, která může zhoršit prognózu nemocných. U tří nemocných jsme prokázali velmi vzácný jev parciální netandémovou duplikaci (PNTD) při cytogenetickém nálezu t(9;11)(p22;q23), t(9;12;11)(p22;p13;q23) a t(11;19)(q23;p13.3). Ve dvou případech byla MLL PTD přerušena sekvencemi partnerského genu MLLT3 (9p22), jednou genem MLLT1 (19p13.3). MLL PNTD maskovanou translokací t(9;11)(p22;q23) popsal u B-ALL také Whitman a kol. (2005). Autoři detekovali parciální duplikaci exonů 2 až 6 přerušenou insercí exonu 9 genu MLLT3. Prokázali, že komplexní abnormalita MLL genu vedla k vzniku tří transkriptů: transkriptu MLL PNTD, transkriptu MLL PTD a fúzního transkriptu analogického k transcriptu jednoduché translokace t(9;11)(p22;q23). Přestože zatím zůstává mnoho nezodpovězených otázek ohledně mechanizmu vzniku MLL PNTD, je podle přerušené MLL duplikace jasné, že prvotním mechanizmem je translokace. Poté následuje parciální tandemová duplikace vzniklého fúzního genu. 75
76 Zatímco Whitman a kol. (2005) popsal MLL PNTD v buňkách kostní dřeně u nemocných při prvním a druhém relapsu a předpokládal tak určitou možnou souvislost, my jsme u všech tří nemocných MLL PNTD detekovali při diagnóze AML. MLL PNTD u nemocných č. 19 a 23 jsme spolu s dalšími dvěma zde neuváděnými případy také publikovali (Sarova a kol. 2009). V dětském souboru nemocných s nově diagnostikovanou AML jsme potvrdili značně vyšší incidenci změn chromosomu 11, konkrétně MLL přestaveb (36 %), oproti dospělým nemocným. Prokázali jsme jednoznačnou spojitost s AML FAB subtypem M5. Žádné jiné abnormality chromosomu 11 jsme u dětí neprokázali. Vzhledem ke krátkému časovému intervalu a malému souboru nemocných však nelze prognózu jednotlivých pacientů porovnávat. V naší studii jsme prokázali jednoznačný přínos FISH analýzy a mapování pomocí BAC sond pro vytypování a identifikaci afektovaných oblastí a genů na chromosomu 11. Nevýhodou metody je nutnost velkého množství vstupního materiálu, přítomnost dělících se buněk a nízká sensitivita, pohybující se na úrovni několika desítek kilobází. Někteří autoři proto doporučují k mapování postižených genů užití mikroarray metody (Manning a kol. 2010, De Braekeleer a kol. 2011). SNP array jsme proto použili u 10 nemocných. U čtyř pacientů tak byly odhaleny nové kryptické delece, v jednom případě však abnormalita nebyla metodou FISH potvrzena. U čtyř nemocných byl zlom přehodnocen do jiného chromosomového pruhu než určila metoda mband. Potvrdili jsme tak přínos metody SNP array pro detekci alterovaných oblastí nebalancovaných chromosomových aberací. Detekce chromosomových aberací u pacientů s leukémií je v současné době nezbytná nejen pro identifikaci postižených oblastí genomu, ale také z důvodu stratifikace nemocných do konkrétních prognostických skupin, na jejichž základě je volena léčebná terapie a indikace k transplantaci kostní dřeně. Klasická cytogenetická analýza je však často limitována faktory jako je množství dělících se buněk, míra kvality chromosomů a velikost nádorového klonu nebo chromosomové změny. V těchto případech je FISH vyšetření vhodným doplněním. Proto jsme se také zaměřili na vyhodnocení významu skríningového FISH vyšetření u prvních odběrů buněk kostní dřeně/periferní krve nemocných s nově diagnostikovanou AML, jehož cílem byl průkaz přítomnosti nejčastěji se vyskytujících chromosomových změn u AML, to znamená abnormalit genu MLL (11q23.3), delecí dlouhých ramen chromosomu 5 a 7 a početních změn chromosomu 8. Potvrdili jsme 76
77 značný přínos skríningového vyšetření především u nemocných s nedostatkem dělících se buněk, u nichž FISH analýza byla jediným cytogenetickým informativním přístupem. Pro odhalení kryptických přestaveb genu MLL se FISH metoda ukázala nezbytnou. Kryptický charakter a únik některých translokací MLL genu rutinním cytogenetickým analýzám potvrdili také Park a kol. (2008) a Kim a kol. (2002). Na nutnost systematické detekce MLL přestaveb upozorňují i další autoři (Schoch a kol. 2003, Arnaud a kol. 2005). Abnormality chromosomů 5, 7 a 8 jsme často detekovali jako součást komplexního karyotypu. FISH skríningové vyšetření se také ukázalo velmi přínosné jako rychlá analýza přítomnosti patologických nálezů spojených s nepříznivou prognózou. 77
78 7. ZÁVĚR Určení a charakteristika chromosomových změn u hematologických onemocnění umožňuje lékařům stratifikovat nemocné do specifických skupin definovaných podle společných klinických znaků. Na jejich základě je pak volena léčebná terapie a indikace k transplantaci kostní dřeně. Studium a identifikace chromosomových změn a zlomových míst u hematologických malignit jsou nezbytné i pro identifikaci genů, které asociují se vznikem a progresí onemocnění a které mohou být také důvodem lékové rezistence. Molekulární a funkční analýzy těchto genů a jejich produktů jsou podkladem pro vývoj nových léků a individuální terapie pro nemocného. V současné době patří detekce chromosomových aberací k standardním vyšetřením pacientů s leukémií. Klasická cytogenetická analýza je však limitována faktory jako je množství dělících se buněk, míra kvality chromosomů a velikost nádorového klonu/chromosomové změny. V těchto případech je FISH vyšetření vhodným a nutným doplněním pro stanovení přesné diagnózy. Proto jsme se zaměřili na vyhodnocení skríningového FISH vyšetření u prvních odběrů nemocných s AML (to znamená při diagnóze). Dále jsme identifikovali a charakterizovali změny chromosomu 11 u akutní myeloidní leukémie, a to z pohledu cytogenetického i klinického. V průběhu let 2006 až 2012 jsme v cytogenetické laboratoři ÚHKT a Centru nádorové cytogenetiky vyšetřili 300 dospělých nemocných a 14 dětí s nově diagnostikovanou AML. Změnu chromosomu 11 jsme prokázali v buňkách kostní dřeně, případně periferní krve, u 18 % dospělých a 36 % dětí s AML. Jednotlivé cíle práce jsou zhodnoceny v následujícíh bodech. Prokázali jsme, že změny chromosomu 11 jsou často se vyskytujícím cytogenetickým nálezem u nově diagnostikovaných dospělých nemocných s AML. Alterováno bývá především dlouhé rameno chromosomu 11. Značnou nestabilitu chromosomu 11 u AML jsme potvrdili detekcí dvou a více zlomů na jednom homologním chromosomu nebo přítomností zlomu na obou homologních chromosomech
79 Výsledky naší studie potvrzují, že chromosom 11 se může uplatňovat v leukemogenezi jak vlivem balancovaných, tak i nebalancovaných změn s množstvím rekurentních i náhodných zlomových míst. Rekurentní zlom jsme identifikovali na 11q v genu MLL (11q23.3) a na 11p v genu NUP98 (11p15.4). Další potencionálně nenáhodné zlomy jsme určili na krátkém i dlouhém rameni chromosomu 11, v oblastech 11p13, 11p12 a 11q13.2. Poukázali jsme na nové kandidátní geny s možnou úlohou v patogenezi AML. Vyhodnotili jsme nejčastěji deletované a duplikované/amplifikované oblasti a navrhli kandidátní tumor supresorové geny a onkogeny lokalizované v těchto částech chromosomu 11. Potvrdili jsme, že nemocní s přestavbou genu MLL tvoří poměrně homogenní skupinu mladších pacientů (<50 let) diagnostikovaných převážně jako AML M5, M1 nebo M4, charakterizovanou balancovanými změnami. Prokázali jsme významnou spojitost ostatních změn chromosomu 11 se starším věkem nemocných (>60 let), komplexním karyotypem, nebalancovanou změnou (na 11p převážně vlivem ztráty genetického materiálu a na 11q obvykle ziskem) a velmi nepříznivým klinickým průběhem s průměrnou dobou přežití 4 měsíců. Neprokázali jsme významnou spojitost s konkrétním AML FAB subtypem nebo sekundárním typem AML. U dětských pacientů jsme potvrdili vyšší frekvenci změn chromosomu 11 než u dospělých nemocných. Ve všech přídach se jednalo o balancovanou přestavbu genu MLL spojenou s nízkým věkem (kolem 1 roku) a AML FAB subtypem M5. Skríning zaměřený na změny genu MLL, deleci dlouhých ramen chromosomů 5 a 7 a početní odchylky chromosomů 8 a 9 u AML se ukázal velmi přínosný u pacientů s nedostatkem dělících se buněk, jako rychlá analýza přítomnosti patologických nálezů spojených s nepříznivou prognózou a pro detekci 79
80 kryptických přestaveb MLL genu, které bývají často pod rozlišovací schopností klasické cytogenetické analýzy. Prokázali jsme nesporný klinický i genetický význam detekcí změn chromosomu 11. Z klinického hlediska se nám podařilo definovat novou skupinu nemocných, a to se změnami chromosomu 11 nezahrnujícími přestavbu genu MLL. Bez ohledu na lokalizaci nebo mechanizmus vzniku zlomového místa vykazovali nemocní společné klinické znaky jako je vyšší věk při diagnóze, častý relaps a krátká doba přežití. Z genetického hlediska jsme prokázali značnou nestabilitu chromosomu 11 u nemocných s AML s množstvím náhodných i opakujících se zlomů. Určili jsme často afektované oblasti na chromosomu 11 a identifikovali řadu nových kandidátních genů s možnou úlohou v leukemogenezi. Molekulární objasnění translokace t(15;17)(q22;q21), identifikace alterovaného genu RARA (17q21) a následné zavedení cílené terapie pomocí inhibitorů kyseliny retinové pro nemocné s touto translokací vedlo k vlně nadšení a doufání v objev dalších takových markerů, které by změnily status prognosticky nepříznivého onemocnění na prognosticky příznivé, dobře léčitelné jako tomu je u akutní promyelocytární leukémie. Zatím se však nepodařilo tuto snahu u ostatních akutních leukémií naplnit a možnost cílené a individuální terapie pro nemocné je stále velmi daleko. Již 50 let je léčba nemocných s AML založena na kombinaci cytarabinu a antracyklinu. Řada nemocných však neprofituje z této aktuálně používané léčby, jak dokazujeme i v naší studii. Vývoj nových terapeutik pro léčbu AML je tedy nezbytný. Základem a prvním krokem pro vznik cílené terapie je identifikace alterovaných chromosomových oblastí a konkrétních alterovaných genů. K těm se přiblížíme právě studiem chromosomových změn. 80
81 8. SEZNAM CITOVANÉ LITERATURY 1. Adam Z, Krejčí M, Vorlíček J a kolektiv (2008): Hematologie (2. doplněné a zcela přepracované vydání). Grada. 2. Andersen MK, Christiansen DH, Kirchhoff M, Pedersen-Bjergaard J (2001): Duplication or amplification of chromosome band 11q23, including the unrearranged MLL gene, is recurrent abnormality in therapy-related MDS and AML, and is closely related to mutation of the TP53 gene and to previous therapy with alkylating agents. Genes, chromosomes & cancer, 31: Aplan PD (2006): Chromosomal translocations involving the MLL gene: molecular mechanisms. DNA Repair, 5(9-10): Arnaud B, Douet-Guilbert N, Morel F, Le Bris MJ, Herry A, Banzakour S, Bourquard P, Morice P, Calvez G, Marion V, Abgrall J, Berthou Ch, Braekeleer M (2005): Screening by fluorescence in situ hybridization for MLL status at diagnosis in 239 unselected patients with acute myeloblastic leukemia. Cancer Genet Cytogenet 161: Aventín A, La Starza R, Casas S, Nomdedéu J, Queipo de Llano MP, Cimino G, Lo Coco F, Sierra J, Mecucci C (2003): MLL tandem duplication in two cases of acute myelocytic leukemia with unbalanced translocations: der(16)t(11;16)(q23;p13) and der(18)t(11;18)(q22;p11.2). Cancer Genet Cytogenet,142(1): Ayton P, Cleary M (2001): Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene, 20: Babicka L, Ransdorfova S, Brezinova J, Zemanova Z, Sindelarova L, Siskova M, Maaloufova J, Cermak J, Michalova K (2007): Analysis of complex chromosomal rearrangements in adult patients with MDS and AML by multicolor FISH. Leuk Res, 31(1): Balgobind BV, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, Beverloo HB, Chang M, Creutzig U, Dworzak MN, Forestier E, Gibson B, Hasle H, Harrison CJ, Heerema NA, Kaspers GJ, Leszl A, Litvinko N, Nigro LL, Morimoto A, Perot C, Pieters R, Reinhardt D, Rubnitz JE, Smith FO, Stary J, Stasevich I, Strehl S, Taga T, Tomizawa D, Webb D, Zemanova Z, Zwaan CM, van den Heuvel-Eibrink MM (2009): Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood, 114(12): Basecke J, Podleschny M, Clemens R, Schnittger S, Viereck V, Trumper L, Griesinger F (2006): Lifelong persistence of AML associated MLL partial tandem duplications (MLL-PTD) in healthy adults. Leuk Res, 30(9): Bloomfield CD, Lawrence D, Byrd JC, Carroll A, Pettenati MJ, Tantravahi R, Patil SR, Davey FR, Berg DT, Schiffer CA, Arthur DC, Mayer RJ (1998): Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res, 58(18):
82 11. Borrow J, Shearman AM, Stanton VP Jr, Becher R, Collins T, Williams AJ, Dube I, Katz F, Knowng YL, Morris C, Ohiashiki K, Toyama K, Rowley J, Housman DE (1996): The t(7;11)(p15;p13) translocation in acute myeloid leukemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet 12: Boultwood J, Pellagatti A, McKenzie AN, Wainscoat JS (2010): Advances in the 5q- syndrome. Blood, 116(26): Braekeleer M, Morel F, Bris M-J, Herry A, Douget-guilbert N (2005): The MLL gene and translocations involving chromosomal band 11q23 in acute leukemia. Anticancer research, 25: Brezinová J, Zemanová Z, Cermák J, Kurková S, Sindelárová L, Schwarz J, Michalová K (2002): Variations in MLL amplification in a patient with acute myeloid leukemia. Leuk Lymphoma, 43(10): Brezinová J, Zemanová Z, Ransdorfová S, Pavlistová L, Babická L, Housková L, Melichercíková J, Sisková M, Cermák J, Michalová K (2007): Structural aberrations of chromosome 7 revealed by a combination of molecular cytogenetic techniques in myeloid malignancies. Cancer Genet Cytogenet, 173(1): Caligiuri MA, Strout MP, Lawrence D, Arthur DC, Baer MR, Yu F, Knuutila S, Mrózek K, Oberkircher AR, Marcucci G, de la Chapelle A, Elonen E, Block AW, Rao PN, Herzig GP, Powell BL, Ruutu T, Schiffer CA, Bloomfield CD (1998): Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res, 58(1): Caligiuri MA, Strout MP, Schichman SA, Mrózek K, Arthur DC, Herzig GP, Baer MR, Schiffer CA, Heinonen K, Knuutila S, Nousiainen T, Ruutu T, Block AW, Schulman P, Pedersen-Bjergaard J, Croce CM, Bloomfield CD (1996): Partial tandem duplication of ALL1 as a recurrent molecular defect in acute myeloid leukemia with trisomy 11. Cancer Res, 15;56(6): Calin GA, Pekarsky Y, Croce CM (2007): The role of microrna and other non-coding RNA in the pathogenesis of chronic lymphocytic leukemia. Best Pract Res Clin Haematol, 20(3): Casas S, Aventín A, Nomdedéu J, Sierra J (2003): Cryptic t(5;11)(q35;p15.5) in adult de novo acute myelocytic leukemia with normal karyotype. Cancer Genet Cytogenet, 145(2): Collins EC, Rabbitts TH (2002): The promiscuous MLL gene links chromosomal translocations to cellular differentiation and tumour tropism. Trends Mol Med, 8(9): Curtiss NP, Bonifas JM, Lauchle JO, Balkman JD, Kratz CP, Emerling BM, Green ED, Le Beau MM, Shannon KM (2005): Isolation and analysis of candidate 82
83 myeloid tumor suppressor genes from a commonly deleted segment of 7q22. Genomics, 85(5): Daser A, Rabbitts TH (2004): Extending the repertoire of the mixed-lineage leukemia gene MLL in leukemogenesis. Genes Dev, 18(9): De Braekeleer E, Meyer C, Douet-Guilbert N, Morel F, Le Bris MJ, Berthou C, Arnaud B, Marschalek R, Férec C, De Braekeleer M (2010): Complex and cryptic chromosomal rearrangements involving the MLL gene in acute leukemia: a study of 7 patients and review of the literature. Blood Cells Mol Dis, 44(4): De Braekeleer E, Douet-Guilbert N, Basinko A, Morel F, Le Bris MJ, Férec C, De Braekeleer M (2011): Using bacterial artificial chromosomes in leukemia research: the experience at the university cytogenetics laboratory in Brest, France. J Biomed Biotechnol Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Löwenberg B, Bloomfield CD; European LeukemiaNet. (2010): Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood, 115(3): Döhner K, Tobis K, Ulrich R, Fröhling S, Benner A, Schlenk RF, Döhner H (2002): Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol, 20(15): Dou Y, Hess JL (2008): Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int J Hematol, 87(1): Douet-Guilbert N, Arnaud B, Morel F, Le Bris MJ, De Braekeleer M (2005): Cryptic 5'MLL gene insertion in an X-chromosome in acute myeloblastic leukemia. Cancer Genet Cytogenet, 157(2): Dyson MJ, Talley PJ, Reilly JT, Stevenson D, Parsons E, Tighe J (2003): Detection of cryptic MLL insertions using a commercial dual-color fluorescence in situ hybridization probe. Cancer Genet Cytogenet, 147(1): Escobar PA, Smith MT, Vasishta A, Hubbard AE, Zhang L (2007): Leukaemia-specific chromosome damage detected by comet with fluorescence in situ hybridization (comet-fish). Mutagenesis, 22(5): Ghanem H, Tank N, Tabbara IA (2012): Prognostic implications of genetic aberrations in acute myelogenous leukemia with normal cytogenetics. Am J Hematol, 87(1): Grimwade D, Hills RK (2009): Independent prognostic factors for AML outcome. Hematology Am Soc Hematol Educ Program,
84 33. Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ, Burnett AK; National Cancer Research Institute Adult Leukaemia Working Group (2010): Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood, 116(3): Grimwade D, Mrózek K (2011): Diagnostic and prognostic value of cytogenetics in acute myeloid leukemia. Hematol Oncol Clin North Am, 25(6): Grimwade D, Vyas P, Freeman S (2010): Assessment of minimal residual disease in acute myeloid leukemia. Curr Opin Oncol, 22(6): Grimwade D, Walker H, Harrison G, Oliver F, Chatters S, Harrison CJ, Wheatley K, Burnett AK, Goldstone AH; Medical Research Council Adult Leukemia Working Party (2001): The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood, 98(5): Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, Goldstone A (1998): The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood, 92(7): Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, Spanhol-Rosseto A, Klein HU, Dugas M, Schindela S, Trifonov V, Schnittger S, Haferlach C, Bassan R, Wells VA, Spinelli O, Chan J, Rossi R, Baldoni S, De Carolis L, Goetze K, Serve H, Peceny R, Kreuzer KA, Oruzio D, Specchia G, Di Raimondo F, Fabbiano F, Sborgia M, Liso A, Farinelli L, Rambaldi A, Pasqualucci L, Rabadan R, Haferlach T, Falini B (2011): Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood, 118(23): Gu Y, Alder H, Nakamura T, Schichman SA, Prasad R, Canaani O, Saito H, Croce CM, Canaani E (1994): Sequence analysis of the breakpoint cluster region in the ALL-1 gene involved in acute leukemia. Cancer Res, 54(9): Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, Croce CM, Canaani E (1992): The t(4;11) chromosome translocation of human acute leukemias fuse the ALL-1 gene, related to Drosophila Thithorax, to the AF-4 gene. Cell, 71: Gué M, Sun J-S, Boudier T (2006): Simultaneous localization of MLL, AF4 and ENL genes in interphase nuclei by 3D-FISH: MLL translocation revisited. BMC Cancer, 6: Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA (2005): Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A, 102(24):
85 43. Gupta M, Raghavan M, Gale RE, Chelala C, Allen C, Molloy G, Chaplin T, Linch DC, Cazier JB, Young BD (2008): Novel regions of acquired uniparental disomy discovered in acute myeloid leukemia. Genes, chromosomes & cancer, 47(9): Haferlach C, Kern W, Schindela S, Kohlmann A, Alpermann T, Schnittger S, Haferlach T (2012): Gene expression of BAALC, CDKN1B, ERG, and MN1 adds independent prognostic information to cytogenetics and molecular mutations in adult acute myeloid leukemia. Genes, chromosomes & cancer, 51(3): Haferlach T, Schoch C, Löffler H, Gassmann W, Kern W, Schnittger S, Fonatsch C, Ludwig WD, Wuchter C, Schlegelberger B, Staib P, Reichle A, Kubica U, Eimermacher H, Balleisen L, Grüneisen A, Haase D, Aul C, Karow J, Lengfelder E, Wörmann B, Heinecke A, Sauerland MC, Büchner T, Hiddemann W (2003): Morphologic dysplasia in de novo acute myeloid leukemia (AML) is related to unfavorable cytogenetics but has no independent prognostic relevance under the conditions of intensive induction therapy: results of a multiparameter analysis from the German AML Cooperative Group studies. J Clin Oncol, 21(2): Hall GW (2001): Childhood myeloid leukaemias. Best Pract Res Clin Haematol, 14(3): Hanahan D, Weinberg RA (2011): Hallmarks of cancer: the next generation. Cell, 144(5): Herry A, Douet-Guilbert N, Gueganic N, Morel F, Le Bris MJ, Berthou C, De Braekeleer M (2006): Del(5q) and MLL amplification in homogeneously staining region in acute myeloblastic leukemia: a recurrent cytogenetic association. Ann Hematol, 85(4): Hesketh R (1997): The Oncogene and Tumour Suppressor Gene, 2nd Ed. Academic Press, London, UK. 50. Hess JL (2004): MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med, 10(10): Hidaka E, Tanaka M, Matsuda K, Ishikawa-Matsumura M, Yamauchi K, Sano K, Honda T, Wakui K, Yanagisawa R, Nakazawa Y, Sakashita K, Shiohara M, Ishii E, Koike K (2007): A complex karyotype, including a three-way translocation generating a NUP98-HOXD13 transcript, in an infant with acute myeloid leukemia. Cancer Genet Cytogenet, 15;176(2): Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, Pratcorona M, Abbas S, Kuipers JE, van Galen JF, Beverloo HB, Sonneveld E, Kaspers GJ, Trka J, Baruchel A, Zimmermann M, Creutzig U, Reinhardt D, Pieters R, Valk PJ, Zwaan CM (2011): NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood, 29;118(13):
86 53. Chen L, Gilkes DM, Pan Y, Lane WS, Chen J (2005): ATM and Chk2- dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J, 24 (19): Cho WC (2007): OncomiRs: the discovery and progress of micrornas in cancers. Mol Cancer, 6: Chowdhury T, Brady HJ (2008): Insights from clinical studies into the role of the MLL gene in infant and childhood leukemia. Blood Cells Mol Dis, 40(2): Ishikawa M, Yagasaki F, Okamura D, Maeda T, Sugahara Y, Jinnai I, Bessho M (2007): A novel gene, ANKRD28 on 3p25, is fused with NUP98 on 11p15 in a cryptic 3-way translocation of t(3;5;11)(p25;q35;p15) in an adult patient with myelodysplastic syndrome/acute myelogenous leukemia. Int J Hematol, 86(3): Iwamoto M, Asakawa H, Hiraoka Y, Haraguchi T (2010): Nucleoporin Nup98: a gatekeeper in the eukaryotic kingdoms. Genes Cells, 15(7): Iyer RV, Sait SN, Matsui S, Block AW, Barcos M, Slack JL, Wetzler M, Baer MR (2004): Massive hyperdiploidy and tetraploidy in acute myelocytic leukemia and myelodysplastic syndrome. Cancer Genet Cytogenet. Jan 1;148(1): Jädersten M, Hellström-Lindberg E (2010): New clues to the molecular pathogenesis of myelodysplastic syndromes. Exp Cell Res 316(8): Jaju RJ, Fidler C, Haas OA, Strickson AJ, Watkins F, Clark K, Cross NC, Cheng JF, Aplan PD, Kearney L, Boultwood J, Wainscoat JS (2001): A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood, 98(4): Jaroslav P, Martina H, Jiri S, Hana K, Petr S, Tomas K, Julius M, Cedrik H (2005): Expression of cyclins D1, D2, and D3 and Ki-67 in Leukemia. Leuk Lymphoma 46 (11): Jarosova M, Takacova S, Holzerova M, Priwitzerova M, Divoka M, Lakoma I, Mihal V, Indrak K, Divoky V (2005): Cryptic MLL-AF10 fusion cause by insertion of duplicated 5 part of MLL into 10p12 in acute leukemia: a case report. Cancer Genet Cytogenet, 162: Jordheim LP, Sève P, Trédan O, Dumontet C (2011): The ribonucleotide reductase large subunit (RRM1) as a predictive factor in patients with cancer. Lancet Oncol, 12(7): Khan I, Malinge S, Crispino J (2011): Myeloid leukemia in Down syndrome. Crit Rev Oncog, 16(1-2): Kim HJ, Cho HI, Kim EC, Ko EK, See CJ, Park SY, Lee DS (2002): A study on 289 consecutive Korean patients with acute leukaemias revealed fluorescence in situ hybridization detects the MLL translocation without cytogenetic evidence both initially and during follow-up. Br J Haematol, 119(4):
87 66. Klaus M, Schnittger S, Haferlach T, Dreyling M, Hiddemann W, Schoch C (2003): Cytogenetics, fluorescence in situ hybridization, and reverse transcriptase polymerase chain reaction are necessary to clarify the various mechanisms leading to an MLL-AF10 fusion in acute myelocytic leukemia with 10;11 rearrangement. Cancer Genet Cytogenet, 144(1): Kobzev YN, Marrtinez-Climent J, Lee S, Chen J, Rowly JD (2004): Analysis of translocations that involve the NUP98 gene in patients with 11p15 chromosomal rearrangements. Genes, Chromosomes & Cancer 41: Kourlas PJ, Strout MP, Becknell B, Veronese ML, Croce CM, Theil KS, Krahe R, Ruutu T, Knuutila S, Bloomfield CD, Caligiuri MA (2000): Identification of a gene at 11q23 encoding a guanine nucleotide exchange factor: evidence for its fusion with MLL in acute myeloid leukemia. Proc Natl Acad Sci U S A, 97(5): La Starza R, Brandimarte L, Pierini V, Nofrini V, Gorello P, Crescenzi B, Berchicci L, Matteucci C, Romoli S, Beacci D, Rosati R, Martelli MF, Mecucci C (2009): A NUP98-positive acute myeloid leukemia with a t(11;12)(p15;q13) without HOXC cluster gene involvement. Cancer Genet Cytogenet, 193(2): Levy V, Ugo V, Delmer A, Tang R, Ramond S, Perrot J, Vrhovac R, Marie JP, Zittoun R, Ajchenbaum-Cymbalista F (1999): Cyclin D1 overexpression allows identification of an aggresive subset of leukemic lymfoproliferative disorder. Leukemia 13(9): Limbergen VH, Poppe B, Michaux L, Herens C, Brown J, Noens L, Berneman Z, De Bock R, De Paepe A, Speleman F (2002): Identification of cytogenetic subclasses and recurring chromosomal aberrations in AML and MDS with complex karyotypes using M-FISH. Genes, chromosomes & cancer, 33(1): Liu H, Cheng EH, Hsieh JJ (2007): Bimodal degradation of MLL by SCFSkp2 and APCCdc20 assures cell cycle execution: a critical regulatory circuit lost in leukemogenic MLL fusions. Genes Dev, 21(19): Luquet I, Laï JL, Barin C, Baranger L, Bilhou-Nabera C, Lippert E, Gervais C, Talmant P, Cornillet-Lefebvre P, Perot C, Nadal N, Mozziconacci MJ, Lafage- Pochitaloff M, Eclache V, Mugneret F, Lefebvre C, Herens C, Speleman F, Poirel H, Tigaud I, Cabrol C, Rousselot P, Daliphard S, Imbert M, Garand R, Geneviève F, Berger R, Terre C (2007): Hyperdiploid karyotypes in acute myeloid leukemia define a novel entity: a study of 38 patients from the Groupe Francophone de Cytogenetique Hematologique (GFCH). Leukemia, 11: Lundin C, Horvat A, Karlsson K, Olofsson T, Paulsson K, Johansson B (2011): t(9;11)(p22;p15) [NUP98/PSIP1] is a poor prognostic marker associated with de novo acute myeloid leukaemia expressing both mature and immature surface antigens. Leuk Res, 35(6):e Manning M, Hudgins L, Professional Practice and Guidelines Committee (2010): Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med, 12(11):
88 76. Manola KN (2009): Cytogenetics of pediatric acute myeloid leukemia. Eur J Haematol, 83(5): Marcucci G, Mrózek K, Ruppert AS, Archer KJ, Pettenati MJ, Heerema NA, Carroll AJ, Koduru PR, Kolitz JE, Sterling LJ, Edwards CG, Anastasi J, Larson RA, Bloomfield CD (2004): Abnormal cytogenetics at date of morphologic complete remission predicts short overall and disease-free survival, and higher relapse rate in adult acute myeloid leukemia: results from cancer and leukemia group B study J Clin Oncol, 22(12): Marchesi F, Annibali O, Cerchiara E, Tirindelli MC, Avvisati G (2011): Cytogenetic abnormalities in adult non-promyelocytic acute myeloid leukemia: a concise review. Crit Rev Oncol Hematol, 80(3): Marschalek R (2011): Mechanisms of leukemogenesis by MLL fusion proteins. Br J Haematol, 152(2): Matsuda K, Hidaka E, Ishida F, Yamauchi K, Makishima H, Ito T, Suzuki T, Imagawa E, Sano K, Katsuyama T, Ota H (2006): A case of acute myelogenous leukemia with MLL-AF10 fusion caused by insertion of 5' MLL into 10p12, with concurrent 3' MLL deletion. Cancer Genet Cytogenet, 171(1): Mayer J, Starý J (2002): Leukemie. 1.vydání, Grada Publishing, Praha. 82. Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, Ben Abdelali R, Macintyre E, De Braekeleer E, De Braekeleer M, Delabesse E, de Oliveira MP, Cavé H, Clappier E, van Dongen JJ, Balgobind BV, van den Heuvel-Eibrink MM, Beverloo HB, Panzer-Grümayer R, Teigler-Schlegel A, Harbott J, Kjeldsen E, Schnittger S, Koehl U, Gruhn B, Heidenreich O, Chan LC, Yip SF, Krzywinski M, Eckert C, Möricke A, Schrappe M, Alonso CN, Schäfer BW, Krauter J, Lee DA, Zur Stadt U, Te Kronnie G, Sutton R, Izraeli S, Trakhtenbrot L, Lo Nigro L, Tsaur G, Fechina L, Szczepanski T, Strehl S, Ilencikova D, Molkentin M, Burmeister T, Dingermann T, Klingebiel T, Marschalek R (2009): New insights to the MLL recombinome of acute leukemias. Leukemia, 23(8): Michaux L, Wlodarska I, Stul M, Dierlamm J, Mugneret F, Herens C, Beverloo B, Verhest A, Verellen-Dumoulin C, Verhoef G, Selleslag D, Madoe V, Lecomte M, Deprijck B, Ferrant a, Delannoy A, Marichal S, Duhem C, Dicato M, Hagemeijer A (2000): MLL amplification in myeloid leukemias: a study of 14 cases with multiple copies of 11q23. Genes, chromosomes & cancer 29: Mitelman F (2000): Recurrent chromosome aberrations in cancer. Mutat Res, 462(2-3): Mrózek K (2008): Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol, 35(4): Mrózek K, Heerema NA, Bloomfield CD (2004): Cytogenetics in acute leukemia. Blood Rev, 18(2):
89 87. Mrózek K, Heinonen K, Lawrence D, Carroll AJ, Koduru PR, Rao KW, Strout MP, Hutchison RE, Moore JO, Mayer RJ, Schiffer CA, Bloomfield CD (1997): Adult patients with de novo acute myeloid leukemia and t(9;11)(p22;q23) have a superior outcome to patients with other translocations involving band 11q23: a cancer and leukemia group B study. Blood, 90(11): Mrózek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD (2007): Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood, 109(2): Nebral K, König M, Schmidt HH, Lutz D, Sperr WR, Kalwak K, Brugger S, Dworzak MN, Haas OA, Strehl S (2005): Screening for NUP98 rearrangements in hematopoietic malignancies by fluorescence in situ hybridization. Haematologica, 90(6): Nemoto N, Suzukawa K, Shimizu S, Shinagawa A, Takei N, Taki T, Hayashi Y, Kojima H, Kawakami Y, Nagasawa T (2007): Identification of a novel fusion gene MLL-MAML2 in secondary acute myelogenous leukemia and myelodysplastic syndrome with inv(11)(q21q23). Genes, chromosomes & cancer, 46(9): Nikitina EG, Urazova LN, Stegny VN (2012): MicroRNAs and human cancer. Exp Oncol, 34(1): Nilson I, Löchner K, Siegler G, Greil J, Beck JD, Fey GH, Marschalek R (1996): Exon/intron structure of the human ALL-1 (MLL) gene involved in translocations to chromosomal region 11q23 and acute leukaemias. Br J Haematol, 93(4): Ng WL, Yan D, Zhang X, Mo YY, Wang Y (2010): Over-expression of mir- 100 is responsible for the low-expression of ATM in the human glioma cell line: M059J. DNA Repair (Amst), 9(11): Nowak N, Shows T (1995): Genetics of chromosome 11: Loci for pediatric and adult malignancies, developmental disorders, and other diseases. Cancer Investigation, 13(6): Pajuelo-Gámez JC, Cervera J, García-Casado Z, Mena-Durán AV, Valencia A, Barragán E, Such E, Bolufer P, Sanz MA (2007): MLL amplification in acute myeloid leukemia. Cancer Genet Cytogenet, 174(2): Panagopoulos I, Fioretos T, Isaksson M, Larsson G, Billström R, Mitelman F, Johansson B (2002): Expression of NUP98/TOP1, but not of TOP1/NUP98, in a treatment-related myelodysplastic syndrome with t(10;20;11)(q24;q11;p15). Genes, chromosomes & cancer, 34(2): Papenhausen PR, Griffin S, Tepperberg J (2005): Oncogene amplification in transforming myelodysplasia. Experimental and Molecular pathology, 79: Park TS, Lee ST, Song J, Lee KA, Lee SG, Kim J, Suh B, Kim SJ, Lee JH, Park R, Choi JR (2008): MLL rearrangement with t(6;11)(q15;q23) as a sole 89
90 abnormality in a patient with de novo acute myeloid leukemia: conventional cytogenetics, FISH, and multicolor FISH analyses for detection of rare MLL-related chromosome abnormalities. Cancer Genet Cytogenet, 187(1): Poirel H, Rack K, Delabesse E, Radford-Weiss I, Troussard X, Debert C, Leboeuf D, Bastard C, Picard F, Veil-Buzyn A, Flandrin G, Bernard O, Macintyre E (1996): Incidence and characterization of MLL gene (11q23) rearrangements in acute myeloid leukemia M1 and M5. Blood, 87(6): Poppe B, Vandesompele J, Schoch C, Lindvall C, Mrozek K, Bloomfield CD, Beverloo HB, Michaux L, Dastugue N, Herens C, Yigit N, De Paepe A, Hagemeijer A, Speleman F (2004): Expression analyses identify MLL as a prominent target of 11q23 amplification and support an etiologic role for MLL gain of function in myeloid malignancies. Blood, 103(1): Qian Z, Joslin JM, Tennant TR, Reshmi SC, Young DJ, Stoddart A, Larson RA, Le Beau MM (2010): Cytogenetic and genetic pathways in therapy-related acute myeloid leukemia. Chem Biol Interact. Mar 19;184(1-2): Rauch I, Kofler B (2010): The galanin system in cancer. EXS, 102: Rowley JD (1999): The role of chromosome translocations in leukemogenesis. Semin Hematol, 36(7): Rowley JD a Olney HJ (2002): International workshop on the relationship of prior therapy to balanced chromosome aberrations in therapy-related myelodysplastic syndromes and acute leukemia: overview report. Genes, chromosomes & cancer 33(4): Rücker FG, Bullinger L, Schwaenen C, Lipka DB, Wessendorf S, Fröhling S, Bentz M, Miller S, Scholl C, Schlenk RF, Radlwimmer B, Kestler HA, Pollack JR, Lichter P, Döhner K, Döhner H (2006): Disclosure of candidate genes in acute myeloid leukemia with complex karyotypes using microarray-based molecular characterization. J Clin Oncol, 24(24): Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, Gaidzik VI, Paschka P, Held G, von Lilienfeld-Toal M, Lübbert M, Fröhling S, Zenz T, Krauter J, Schlegelberger B, Ganser A, Lichter P, Döhner K, Döhner H (2012): TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood, 119(9): Sandberg AA, Meloni-Ehrig AM (2010): Cytogenetics and genetics of human cancer: methods and accomplishments. Cancer Genet Cytogenet, 203(2): Sárová I, Brezinová J, Zemanová Z, Izáková S, Lizcová L, Malinová E, Berková A, Cermák J, Maaloufová J, Nováková L, Michalová K (2010): Cytogenetic manifestation of chromosome 11 duplication/amplification in acute myeloid leukemia. Cancer Genet Cytogenet, 199(2):
91 109. Sárová I, Brezinová J, Zemanová Z, Lizcová L, Berková A, Izáková S, Malinová E, Fuchs O, Kostecka A, Provazníková D, Filkuková J, Maaloufová J, Starý J, Michalová K (2009): A partial nontandem duplication of the MLL gene in four patients with acute myeloid leukemia. Cancer Genet Cytogenet, 195(2): Shaffer LG, Slovak ML, Cambel LJ, editors (2009): ISCN: an international system for human cytogenetic nomenclature. Basel: S. Karger Schnittger S, Kinkelin U, Schoch C, Heinecke A, Haase D, Haferlach T, Büchner T, Wörmann B, Hiddemann W, Griesinger F (2000): Screening for MLL tandem duplication in 387 unselected patients with AML identify a prognostically unfavorable subset of AML. Leukemia, 14(5): Schoch C, Haferlach T, Haase D, Fonatsch C, Loffer H, Schlegelberger B, Staib P, Sauerland MC, Heinecke A, Buchner T, Hiddemann W, German AML Study Group (2001): Patients with de novo acute myeloid leukaemia and complex karyotype aberrations show a poor prognosis despite intensive treatment: a study of 90 patients. Br J Haematol, 112: Schoch C, Schnittger S, Klaus M, Kern W, Hiddemann W, Haferlach T (2003): AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood, 102(7): Slany RK (2005): When epigenetics kills: MLL fusion proteins in leukemia. Hematological Oncology, 23: So CW, Cleary ML (2004): Dimerization: A versatile switch for oncogenesis. Blood, 104: Soukup P, Soukupová Maaloufová J, Čermák J, Cetkovský P, Lukášová M (2012): Akutní myeloidní leukémie-historický vývoj a současnost léčby ve světě a v UHKT. Vnitřní lékařství 58; suplplementum 2S Stilgenbauer S, Schaffner C, Litterst A, Liebisch P, Gilad S, Bar-Shira A, James MR, Lichter P, Dohner H (1997): Biallelic mutations in the ATM gene in T- prolymphocytic leukemia. Nat Med, 3(10): Strissel PL, Strick R, Rowley JD, Zeleznik-Le NJ (1998): An in vivo topoisomerase II cleavage site and a DNase I hypersensitive site colocalize near exon 9 in the MLL breakpoint cluster region. Blood, 92(10): Strout MP, Marcucci G, Bloomfield CD, Caligiuri MA (1998): The partial tandem duplication of ALL1 b(mll) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc Natl Acad Sci USA, 95: Sung PA, Libura J, Richardson C (2006): Etoposide and illegitimate DNA double-strand break repair in the generation of MLL translocations: New insights and new questions. DNA Repair, 5(9-10):
92 121. Swerdlow SH, Campo, E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (2008): WHO Classification of Tumours of Haematopoietic and Lmyphoid Tissues, IARC, Lyon Takáčová S, Jarošová M, Divoký V (2007): Porucha v regulácii chromatínu ako molekulárny mechanizmus MLL-ENL leukemogenézy. Trans.Hemat.dnes, 13: Tanaka K, Takechi M, Nishimura S, Oguma N, Kamada N (1993): Amplification of c-myc oncogene and point mutation of N-RAS oncogene point mutation in acute myelocytic leukemias with double minute chromosomes. Leukemia, 7(3): Taylor TD, Noguchi H, Totoki Y, Toyoda A, Kuroki Y, Dewar K, Lloyd C, Itoh T, Takeda T, Kim DW, She X, Barlow KF, Bloom T, Bruford E, Chang JL, Cuomo CA, Eichler E, FitzGerald MG, Jaffe DB, LaButti K, Nicol R, Park HS, Seaman C, Sougnez C, Yang X, Zimmer AR, Zody MC, Birren BW, Nusbaum C, Fujiyama A, Hattori M, Rogers J, Lander ES, Sakaki Y (2006): Human chromosome 11 DNA sequence and analysis including novel gene identification. Nature, 440(7083): Tennant TR, Huo D, Davis EM, Larson RA, Le Bau MM (2007): Genomic Rearrangements Associated with -5/del(5q) and -7/del(7q) in Myeloid Leukemias. Journal of the american society of hematology, 110(11):537a Vieira L, Marques B, Cavaleiro C, Ambrósio AP, Jorge M, Neto A, Costa JM, Júnior EC, Boavida MG (2005): Molecular cytogenetic characterization of rearrangements involving 12p in leukemia. Cancer Genet Cytogenet, 157(2): Vieira L, Sousa AC, Matos P, Marques B, Alaiz H, Ribeiro MJ, Braga P, da Silva MG, Jordan P (2006): Three-way translocation involves MLL, MLLT3, and a novel cell cycle control gene, FLJ10374, in the pathogenesis of acute myeloid leukemia with t(9;11;19)(p22;q23;p13.3). Genes, chromosomes & cancer, 45(5): Vorechovsky I, Luo L, Dyer MJ, Catovsky D, Amlot PL, Yaxley JC, Foroni L, Hammarstrom L, Webster AD, Yuille MA (1997): Clustering of missense mutations in the ataxia-telangiectasia gene in a sporadic T-cell leukemia. Nat Genet, 17(1): Wang Y, Xue Y, Chen S, Wu Y, Pan J, Zhang J, Shen J (2010): A novel t(5;11)(q31;p15) involving the NUP98 gene on 11p15 is associated with a loss of the EGR1 gene on 5q31 in a patient with acute myeloid leukemia. Cancer Genet Cytogenet, 199(1): Whitman SP, Liu S, Vukosavljevic T, Rush LJ, Yu L, Liu Ch, Klisovic MI, Maharry K, Guimond M, Strout MP, Becknell B, Dorrance A, Klisovic RB, Plass Ch, Bloomfield CD, Marcucci G, Caliguiri MA (2005): The MLL tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood, 1:
93 131. Whitman SP, Strout MP, Marcucci G, Freud AG, Culley LL, Zeleznik-Le NJ, Mrózek K, Theil KS, Kees UR, Bloomfield CD, Caligiuri MA (2001): The partial nontandem duplication of the MLL (ALL1) gene is a novel rearrangement that generates three distinct fusion transcripts in B-cell acute lymphoblastic leukemia. Cancer Res, 61(1): Winrow CJ, Pankratz DG, Vibat CR, Bowen TJ, Callahan MA, Warren AJ, Hilbush BS, Wynshaw-Boris A, Hasel KW, Weaver Z, Lockhart DJ, Barlow C (2005): Aberrant recombination involving the granzyme lokus occurs in Atm-/-T-cell lymphomas. Hum Mol Genet, 14(18): Zatkova A, Merk S, Wendehack M, Bilban M, Muzik EM, Muradyan A, Haferlach C, Haferlach T, Wimmer K, Fonatsch C, Ullmann R (2009): AML/MDS with 11q/MLL amplification show characteristic gene expression signature and interplay of DNA copy number changes. Genes, chromosomes & cancer, 48(6): Zhang L, Alsabeh R, Mecucci C, La Starza R, Gorello P, Lee S, Lill M, Schreck R (2007): Rare t(1;11)(q23;p15) in therapy-related myelodysplastic syndrome evolving into acute myelomonocytic leukemia: a case report and review of the literature. Cancer Genet Cytogenet, 178(1): Zhang Y, Rowley JD (2006): Chromatin structural elements and chromosomal translocations in leukemia. DNA Repair, 5(9-10): Ziemin-van der Poel S, McCabe NR, Gill HJ, Espinosa III R, Pattel YD, Harden AM, Le Beau MM, Smith SB, Rowley JD, Diaz MO (1991): Identification of a gene (MLL) which spans the breakpoint in 11q23 translocations associated with human leukemias. Proc Natl Acad Sci USA, 88:
94 8. SEZNAM PŘÍLOH PŘÍLOHA Č. 1 FAB klasifikace AML PŘÍLOHA Č. 2 WHO klasifikace AML (2008) PŘÍLOHA Č. 3 Rekurentní změny 11p oblast 11p15.4 a 11p13 (nemocný č. 10) PŘÍLOHA Č. 4 Rekurentní změny 11p oblast 11p12 (nemocný č. 13) PŘÍLOHA Č. 5 Rekurentní změny 11q oblast 11q13.2 (nemocný č. 54) PŘÍLOHA Č. 6 Rekurentní změny 11q oblast 11q23.3 (nemocný č. 26) PŘÍLOHA Č. 7 Ukázky různých forem duplikace/amplifikace 94
95 PŘÍLOHA Č. 1 FAB klasifikace AML FAB subtyp Název Výskyt Charakteristika M0 Nediferenciovaná AML 3 % M1 AML bez vyzrávání 15 % M2 AML s vyzráváním 25 % M3 M4 M4eo M5 M6 M7 Akutní promyelocytární leukémie Akutní myelomonocytární leukémie Akutní myelomonocytární leukémie s eozinofilií Akutní monocytární leukémie Akutní erytroblastická leukémie Akutní megakaryocytární leukémie Vysvětlivky: MPO, myeloperoxidáza. 5 % 25 % 6 % 3 % 3 % Velké blasty s hojnou cytoplazmou a nápadnými jadérky, cytochemicky negativní Blasty s kulatým jádrem a menším množstvím cytoplazmy s ojedinělými azurofilními granuly a Auerovými tyčemi, MPO a Sudanová čerň pozitivní Blasty obsahují četná azurofilní granula a Auerovy tyče, MPO a Sudanová čerň pozitivní, věk kolem 30 let, častá t(8;21)(q22;q22) Blasty s kulatým jádrem a jadérky, hojné Auerovy tyče a granula, MPO a Sudanová čerň silně pozitivní, věk let, častá t(15;17)(q22;q21) Blasty se světle šedou, granulovanou cytoplazmou a zprohýbanými jádry, pozitivní nespecifické esterázy Blasty vzhledu myelomonocytární leukémie, eozinofily tvoří 5-10 % buněk dřeně, časté abnormity 16q22 Blasty se zprohýbanými jádry a hojnou, lehce granulovanou cytoplazmou s Auerovými tyčemi, častěji <50 let, abnormality 11q23 Bizardní erytroblasty, často mnohojaderné Mikromegakaryocyty jedno či dvoujaderné, kyselá fosfatáza pozitivní, častá inv(3)(q21q26) 95
96 PŘÍLOHA Č. 2 WHO klasifikace AML (2008) AML s charakteristickými cytogenetickými abnormalitami AML s t(8;21)(q22;q22), RUNX1-RUNX1T1 APL s t(15;17)(q22;q21), PML-RARA AML s abn. eozinofily v kostní dřeni a t(16;16)(p13;q22), inv(16)(p13;q22), CBFB-MYH11 AML s t(9;11)(p22;q23), MLLT3-MLL AML s t(6;9)(p23;q34), DEK-NUP214 AML s inv(3)(q21q26.2), t(3;3)(q21;q26.2), RPN1-EVI1 AML s t(1;22)(p13;q13), RBM15-MKL1 AML s mutací NPM1 AML s mutací CEBPA AML s myelodysplastickými změnami Sekundární AML AML blíže nespecifikované AML s minimální diferenciací (AML M0) AML bez vyzrávání (AML M1) AML s vyzráváním (AML M2) Akutní myelomonocytární leukémie (AML M4) Akutní monoblastická/monocytární leukémie (AML M5a/b) Akutní erytroblastická leukémie (AML M6) Akutní megakaryocytární leukémie (AML M7) Akutní bazofilní leukémie Akutní panmyelóza s myelofibrózou Myeloidní sarkom Myeloidní proliferace se vztahem k Downovu syndromu Transientní abnormální myelopoéza Myeloidní leukémie asociovaná s Downovým syndromem Neoplásie blastických plasmocytoidních dendritických buněk 96
 Ideogram komplexního karyotypu 46,XY,der(5)t(5;20)(q35;p?")
,der(20)t(11;20)(p15;p?)t(11;20)(p11.2;q?) (vlevo).")
.")
(")
 prokázala kryptickou deleci chromosomu 11")
 a RP11-265K23 (5q35) prokazující fúzi genů NUP98 a NSD1 (uprostřed")
 a 20q (ToTel 20q) v")
 prokázala balancovaný charakter translokace t(11;20) a přestavbu")
97 PŘÍLOHA Č. 3 Rekurentní změny 11p oblast 11p15.4 a 11p13 (nemocný č. 10) Ideogram komplexního karyotypu 46,XY,der(5)t(5;20)(q35;p?),der(11)t(5;11)(q35;p15),der(11)t(11;20) (p11.2;q?),der(20)t(11;20)(p15;p?)t(11;20)(p11.2;q?) (vlevo). mband analýza s XCyte11 sondou prokazující pouze jedem zlom na chromosomu 11, a to v oblasti 11p11.2 (vlevo dole). Vpravo výsledek mfish analýzy zachycující pouze zdánlivě nebalancovanou translokaci t(11;20)(p12;q?). SNP mikroarray (vlevo) prokázala kryptickou deleci chromosomu 11 oblasti 11p13. FISH se sondami RP11-340A20 (11p15.4) a RP11-265K23 (5q35) prokazující fúzi genů NUP98 a NSD1 (uprostřed nahoře). FISH se subtelomerickou sondou pro 20p (ToTel 20p) a 20q (ToTel 20q) v kombinaci s centromerickou sondou CEP 20 (20p11.1-q11.1) prokázala balancovaný charakter translokace t(11;20) a přestavbu obou ramen chromosomu 20 (vlevo nahoře). FISH s RP11-140L23 (11p13) a CEP 11 (11p11.1-q11.1) potvrdila deleci 11p13 (slabší orandžový signál na chromosomu 11, uprostřed dole). FISH s RP11-433B4 (11p12) a CEP 11 prokazující zlom v oblasti 11p12 (orandžový signál přítomný na der(20) i der(11) (vpravo dole)). 97
 Výsledek mfish analýzy prokazující komplexní karyotyp")
t(7;11)(q22;p13),del(11)(p13),+idic(11)(p11.")
del(18)(q21) [cp22] (vpravo).")
.")
 potvrdila zmnožení oblasti 11p11.")
 (uprostřed nahoře) a RP11-12C11 (11p11.")
 (vpravo nahoře) specifikující zlomové místo do oblasti mezi")
98 PŘÍLOHA Č. 4 Rekurentní změny 11p oblast 11p12 (nemocný č. 13) Výsledek mfish analýzy prokazující komplexní karyotyp 47,XY,der(2)t(2;15)(q34;q24),del(5)(q13q31), der(7)t(7;11)(q22;p13),del(11)(p13),+idic(11)(p11.1),del(15)(q24),der(18)del(18)(p11.2)del(18)(q21) [cp22] (vpravo). mband analýza s XCyte11 sondou odhalila zlomy v oblastech 11p13 a 11p11.1 (vlevo dole). Vlevo nahoře ideogram translokace. SNP mikroarray (vlevo) potvrdila zmnožení oblasti 11p qter. FISH se sondami RP11-450F4 (11p11.2) a CEP 11 (11p11.1-q11.1) (uprostřed nahoře) a RP11-12C11 (11p11.2) a CEP 11 (11p11.1- q11.1) (vpravo nahoře) specifikující zlomové místo do oblasti mezi těmito sondami. FISH se sondami RP11-233I10 (11p12) a CEP 11 (11p11.1-q11.1) (uprostřed dole) a RP11-456C16 (11p12) a CEP 11 (11p11.1-q11.1) (vpravo dole) upřesňují zlomové místo do oblasti mezi těmito sondami. 98

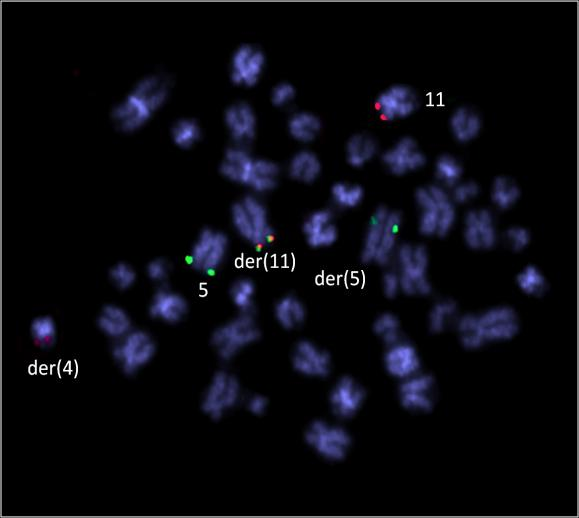
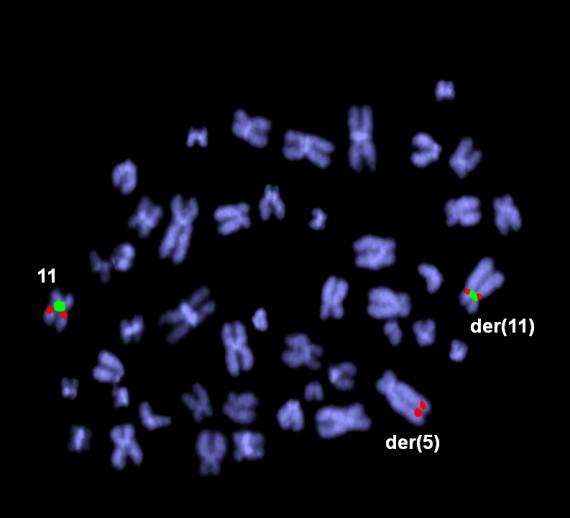
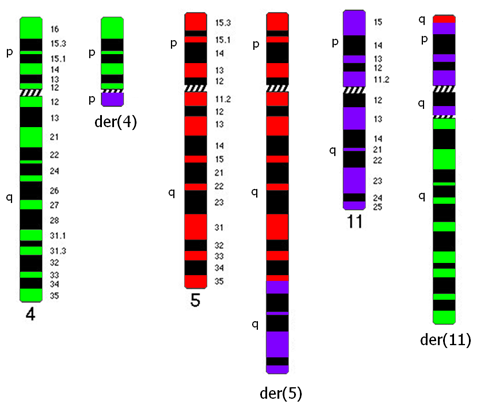
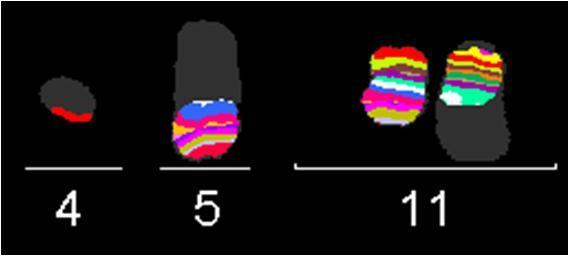

99 PŘÍLOHA Č. 5 Rekurentní změny 11q oblast 11q13.2 (nemocný č. 54) Komplexní karyotyp 46,XY,der(4)t(4;11)(q11;p15),der(5)t(5;11)(q35;q13),der(11)t(4;11)(q11;q13) t(5;11)(q35;p15)[21]: ideogram (vlevo), klasická cytogenetická analýza (vpravo). Zlomy v oblastech 11p15, 11q13.5 a 4q11 určené metodou mband pro chromosom 11 (vlevo nahoře) a chromosom 4 (vlevo dole). Komplexní karyotyp detekovaný metodou mfish (vpravo). FISH se sondami RP11-340A20 (11p15.4) a RP11-265K23 (5q35) prokazující fúzi genů NUP98 a NSD1 (vlevo). FISH s RP11-569N5 (11q13.2) a CEP 11 (11p11.1-q11.1) prokazující zlom v oblasti 11q13.2 (orandžový signál přítomný na der(5) i der(11)) (vpravo). 99
,+4,+8,+8,+12,+16,+21[cp12] prokázaný metodou klasické cytogenetické analýzy (vlevo) a metodou mfish (vpravo).")
 identifikovala zlom v genu MLL (oddělený orandžový a zelený signál) (vlevo).")
 a")
100 PŘÍLOHA Č. 6 Rekurentní změny 11q oblast 11q23.3 (nemocný č. 26) Karyotyp 46~52,XY,t(2;11)(p24;q23.3),+4,+8,+8,+12,+16,+21[cp12] prokázaný metodou klasické cytogenetické analýzy (vlevo) a metodou mfish (vpravo). FISH s dvoubarevnou sondou pro MLL gen (11q23.3) identifikovala zlom v genu MLL (oddělený orandžový a zelený signál) (vlevo). Partnerský chromosom morfologicky neodpovídal alterovanému chromosomu 2, ale zdánlivě normálnímu chromosomu 10. FISH analýza s dvoubarevnou sondou pro MLL gen v kombinaci se subtelomerickými sondami pro krátká (ToTel 10p) a dlouhá ramena (ToTel 10q) chromosomu 10 (vpravo) naši hypotézu inzerce části MLL genu do derivovaného chromosomu 10 potvrdila. 100

101 PŘÍLOHA Č. 7 Ukázky různých forem duplikace/amplifikace 101
Cytogenetické vyšetřovací metody v onkohematologii Zuzana Zemanová
 Cytogenetické vyšetřovací metody v onkohematologii Zuzana Zemanová Centrum nádorové cytogenetiky Ústav klinické biochemie a laboratorní diagnostiky VFN a 1. LF UK v Praze Klinický význam cytogenetických
Cytogenetické vyšetřovací metody v onkohematologii Zuzana Zemanová Centrum nádorové cytogenetiky Ústav klinické biochemie a laboratorní diagnostiky VFN a 1. LF UK v Praze Klinický význam cytogenetických
Akutní leukémie a myelodysplastický syndrom. Hemato-onkologická klinika FN a LF UP Olomouc
 Akutní leukémie a myelodysplastický syndrom Hemato-onkologická klinika FN a LF UP Olomouc Akutní leukémie (AL) Představují heterogenní skupinu chorob charakterizovaných kumulací klonu nevyzrálých, nádorově
Akutní leukémie a myelodysplastický syndrom Hemato-onkologická klinika FN a LF UP Olomouc Akutní leukémie (AL) Představují heterogenní skupinu chorob charakterizovaných kumulací klonu nevyzrálých, nádorově
HODNOCENÍ CYTOGENETICKÝCH A FISH NÁLEZŮ U NEMOCNÝCH S MNOHOČETNÝM MYELOMEM VE STUDII CMG ORGANIZACE VÝZKUMNÉHO GRANTU NR/
 HODNOCENÍ CYTOGENETICKÝCH A FISH NÁLEZŮ U NEMOCNÝCH S MNOHOČETNÝM MYELOMEM VE STUDII CMG 22. ORGANIZACE VÝZKUMNÉHO GRANTU NR/8183-4. KLONÁLNÍ CHROMOSOMOVÉ ABERACE U MNOHOČETNÉHO MYELOMU (MM). Klasické
HODNOCENÍ CYTOGENETICKÝCH A FISH NÁLEZŮ U NEMOCNÝCH S MNOHOČETNÝM MYELOMEM VE STUDII CMG 22. ORGANIZACE VÝZKUMNÉHO GRANTU NR/8183-4. KLONÁLNÍ CHROMOSOMOVÉ ABERACE U MNOHOČETNÉHO MYELOMU (MM). Klasické
Jaké máme leukémie? Akutní myeloidní leukémie (AML) Akutní lymfoblastická leukémie (ALL) Chronické leukémie, myelodysplastický syndrom,
 Akutní lymfoblastická leukémie (ALL) Chronické leukémie, myelodysplastický syndrom,") Akutní myeloidní leukémie (AML) Jaké máme leukémie? Akutní lymfoblastická leukémie (ALL) Chronické leukémie, myelodysplastický syndrom, Chronické leukémie, mnohočetný myelom, Někdy to není tak jednoznačné
Akutní myeloidní leukémie (AML) Jaké máme leukémie? Akutní lymfoblastická leukémie (ALL) Chronické leukémie, myelodysplastický syndrom, Chronické leukémie, mnohočetný myelom, Někdy to není tak jednoznačné
UNIVERZITA KARLOVA V PRAZE Přírodovědecká fakulta Katedra genetiky a mikrobiologie
 UNIVERZITA KARLOVA V PRAZE Přírodovědecká fakulta Katedra genetiky a mikrobiologie Význam aberací MLL genu u nemocných s akutní myeloidní leukemií Iveta Šárová Praha, 2008 Diplomová práce byla vypracována
UNIVERZITA KARLOVA V PRAZE Přírodovědecká fakulta Katedra genetiky a mikrobiologie Význam aberací MLL genu u nemocných s akutní myeloidní leukemií Iveta Šárová Praha, 2008 Diplomová práce byla vypracována
ONKOGENETIKA. Spojuje: - lékařskou genetiku. - buněčnou biologii. - molekulární biologii. - cytogenetiku. - virologii
 ONKOGENETIKA Spojuje: - lékařskou genetiku - buněčnou biologii - molekulární biologii - cytogenetiku - virologii Důležitost spolupráce různých specialistů při detekci hereditárních forem nádorů - (onkologů,internistů,chirurgů,kožních
ONKOGENETIKA Spojuje: - lékařskou genetiku - buněčnou biologii - molekulární biologii - cytogenetiku - virologii Důležitost spolupráce různých specialistů při detekci hereditárních forem nádorů - (onkologů,internistů,chirurgů,kožních
Beličková 1, J Veselá 1, E Stará 1, Z Zemanová 2, A Jonášová 2, J Čermák 1
 Beličková 1, J Veselá 1, E Stará 1, Z Zemanová 2, A Jonášová 2, J Čermák 1 1 Ústav hematologie a krevní transfuze, Praha 2 Všeobecná fakultní nemocnice, Praha MDS Myelodysplastický syndrom (MDS) je heterogenní
Beličková 1, J Veselá 1, E Stará 1, Z Zemanová 2, A Jonášová 2, J Čermák 1 1 Ústav hematologie a krevní transfuze, Praha 2 Všeobecná fakultní nemocnice, Praha MDS Myelodysplastický syndrom (MDS) je heterogenní
Vytvořilo Oddělení lékařské genetiky FN Brno
 GONOSOMY GONOSOMY CHROMOSOMY X, Y Obr. 1 (Nussbaum, 2004) autosomy v chromosomovém páru homologní po celé délce chromosomů crossingover MEIÓZA Obr. 2 (Nussbaum, 2004) GONOSOMY CHROMOSOMY X, Y ODLIŠNOSTI
GONOSOMY GONOSOMY CHROMOSOMY X, Y Obr. 1 (Nussbaum, 2004) autosomy v chromosomovém páru homologní po celé délce chromosomů crossingover MEIÓZA Obr. 2 (Nussbaum, 2004) GONOSOMY CHROMOSOMY X, Y ODLIŠNOSTI
VNL. Onemocnění bílé krevní řady
 VNL Onemocnění bílé krevní řady Změny leukocytů V počtu leukocytů Ve vzájemném zastoupení morfologických typů leukocytů Ve funkci leukocytů Reaktivní změny leukocytů Leukocytóza: při bakteriální infekci
VNL Onemocnění bílé krevní řady Změny leukocytů V počtu leukocytů Ve vzájemném zastoupení morfologických typů leukocytů Ve funkci leukocytů Reaktivní změny leukocytů Leukocytóza: při bakteriální infekci
Cytogenetika. chromosom jádro. telomera. centomera. telomera. buňka. histony. páry bazí. dvoušroubovice DNA
 Cytogenetika telomera chromosom jádro centomera telomera buňka histony páry bazí dvoušroubovice DNA Typy chromosomů Karyotyp člověka 46 chromosomů 22 párů autosomů (1-22 od největšího po nejmenší) 1 pár
Cytogenetika telomera chromosom jádro centomera telomera buňka histony páry bazí dvoušroubovice DNA Typy chromosomů Karyotyp člověka 46 chromosomů 22 párů autosomů (1-22 od největšího po nejmenší) 1 pár
Seznam vyšetření. Nesrážlivá periferní krev nebo kostní dřeň
 VD.PCE 08 Laboratorní příručka Příloha č. 1: Seznam vyšetření Molekulární hematologie a hematoonkologie Vyšetření mutačního stavu IgV H u CLL Detekce přestavby IgV H u jiných B lymfoproliferací RNA nebo
VD.PCE 08 Laboratorní příručka Příloha č. 1: Seznam vyšetření Molekulární hematologie a hematoonkologie Vyšetření mutačního stavu IgV H u CLL Detekce přestavby IgV H u jiných B lymfoproliferací RNA nebo
Seznam vyšetření. Detekce markerů: F2 (protrombin) G20210A, F5 Leiden (G1691A), MTHFR C677T, MTHFR A1298C, PAI-1 4G/5G, F5 Cambridge a Hong Kong
 G20210A, F5 Leiden (G1691A), MTHFR C677T, MTHFR A1298C, PAI-1 4G/5G, F5 Cambridge a Hong Kong") VD.PCE 02 Laboratorní příručka Příloha č. 2: Seznam vyšetření Molekulární hematologie a hematoonkologie Detekce markerů: F2 (protrombin) G20210A, F5 Leiden (G1691A), MTHFR C677T, MTHFR A1298C, PAI-1 4G/5G,
VD.PCE 02 Laboratorní příručka Příloha č. 2: Seznam vyšetření Molekulární hematologie a hematoonkologie Detekce markerů: F2 (protrombin) G20210A, F5 Leiden (G1691A), MTHFR C677T, MTHFR A1298C, PAI-1 4G/5G,
Leukemie a myeloproliferativní onemocnění
 Leukemie a myeloproliferativní onemocnění Myeloproliferativní tumory Klonální onemocnění hematopoetických stem cells charakterizované proliferací jedné nebo více myeloidních řad. Dospělí peak 5. 7. dekáda,
Leukemie a myeloproliferativní onemocnění Myeloproliferativní tumory Klonální onemocnění hematopoetických stem cells charakterizované proliferací jedné nebo více myeloidních řad. Dospělí peak 5. 7. dekáda,
VÝSLEDKY FISH ANALÝZY U NEMOCNÝCH S MM ZAŘAZENÝCH VE STUDII CMG 2002 VÝZKUMNÝ GRANT NR/8183-4 CMG CZECH GROUP M Y E L O M A Č ESKÁ MYELOMOVÁ SKUPINA
 VÝSLEDKY FISH ANALÝZY U NEMOCNÝCH S MM ZAŘAZENÝCH VE STUDII CMG 2002 VÝZKUMNÝ GRANT NR/8183-4 CZECH CMG M Y E L O M A GROUP Č ESKÁ MYELOMOVÁ SKUPINA Zhodnocení spolupráce Přehled molekulárně cytogenetických
VÝSLEDKY FISH ANALÝZY U NEMOCNÝCH S MM ZAŘAZENÝCH VE STUDII CMG 2002 VÝZKUMNÝ GRANT NR/8183-4 CZECH CMG M Y E L O M A GROUP Č ESKÁ MYELOMOVÁ SKUPINA Zhodnocení spolupráce Přehled molekulárně cytogenetických
Atestace z lékařské genetiky inovované otázky pro rok A) Molekulární genetika
 Molekulární genetika") Atestace z lékařské genetiky inovované otázky pro rok 2017 A) Molekulární genetika 1. Struktura lidského genu, nomenklatura genů, databáze týkající se klinického dopadu variace v jednotlivých genech. 2.
Atestace z lékařské genetiky inovované otázky pro rok 2017 A) Molekulární genetika 1. Struktura lidského genu, nomenklatura genů, databáze týkající se klinického dopadu variace v jednotlivých genech. 2.
Molekulární hematologie a hematoonkologie
 VD.PCE 02 Laboratorní příručka Příloha č. 2: Seznam vyšetření GENETIKA Molekulární hematologie a hematoonkologie Detekce markerů: F2 (protrombin) G20210A, F5 Leiden (G1691A F2 (protrombin) G20210A, F5
VD.PCE 02 Laboratorní příručka Příloha č. 2: Seznam vyšetření GENETIKA Molekulární hematologie a hematoonkologie Detekce markerů: F2 (protrombin) G20210A, F5 Leiden (G1691A F2 (protrombin) G20210A, F5
BUNĚČNÁ TRANSFORMACE A NÁDOROVÉ BUŇKY
 BUNĚČNÁ TRANSFORMACE A NÁDOROVÉ BUŇKY 1 VÝZNAM BUNĚČNÉ TRANSFORMACE V MEDICÍNĚ Příklad: Buněčná transformace: postupná kumulace genetických změn Nádorové onemocnění: kolorektální karcinom 2 3 BUNĚČNÁ TRANSFORMACE
BUNĚČNÁ TRANSFORMACE A NÁDOROVÉ BUŇKY 1 VÝZNAM BUNĚČNÉ TRANSFORMACE V MEDICÍNĚ Příklad: Buněčná transformace: postupná kumulace genetických změn Nádorové onemocnění: kolorektální karcinom 2 3 BUNĚČNÁ TRANSFORMACE
Sondy k detekci aneuploidií a mikrodelečních syndromů pro prenatální i postnatální vyšetření
 Sondy k detekci aneuploidií a mikrodelečních syndromů pro prenatální i postnatální vyšetření Název sondy / vyšetřovaného syndromu vyšetřovaný gen / oblast použití Fast FISH souprava prenatálních sond DiGeorge
Sondy k detekci aneuploidií a mikrodelečních syndromů pro prenatální i postnatální vyšetření Název sondy / vyšetřovaného syndromu vyšetřovaný gen / oblast použití Fast FISH souprava prenatálních sond DiGeorge
Sylabus témat ke zkoušce z lékařské biologie a genetiky. Struktura, reprodukce a rekombinace virů (DNA viry, RNA viry), význam v medicíně
, význam v medicíně") Sylabus témat ke zkoušce z lékařské biologie a genetiky Buněčná podstata reprodukce a dědičnosti Struktura a funkce prokaryot Struktura, reprodukce a rekombinace virů (DNA viry, RNA viry), význam v medicíně
Sylabus témat ke zkoušce z lékařské biologie a genetiky Buněčná podstata reprodukce a dědičnosti Struktura a funkce prokaryot Struktura, reprodukce a rekombinace virů (DNA viry, RNA viry), význam v medicíně
Mgr. Veronika Peňásová vpenasova@fnbrno.cz Laboratoř molekulární diagnostiky, OLG FN Brno Klinika dětské onkologie, FN Brno
 Retinoblastom Mgr. Veronika Peňásová vpenasova@fnbrno.cz Laboratoř molekulární diagnostiky, OLG FN Brno Klinika dětské onkologie, FN Brno Retinoblastom (RBL) zhoubný nádor oka, pocházející z primitivních
Retinoblastom Mgr. Veronika Peňásová vpenasova@fnbrno.cz Laboratoř molekulární diagnostiky, OLG FN Brno Klinika dětské onkologie, FN Brno Retinoblastom (RBL) zhoubný nádor oka, pocházející z primitivních
Nestabilita genomu nádorových n buněk mutace a genové či i chromosomové aberace jedna z nejdůle ležitějších událost lostí při i vzniku maligního proce
 MOLEKULÁRNĚ CYTOGENETICKÁ ANALÝZA BUNĚK K MALIGNÍCH MOZKOVÝCH TUMORŮ Zemanová Z. 1, Babická L. 1, Kramář F. 3, Ransdorfová Š. 2, Pavlištov tová L. 1, BřezinovB ezinová J. 2, Hrabal P. 4, Kozler P. 3, Michalová
MOLEKULÁRNĚ CYTOGENETICKÁ ANALÝZA BUNĚK K MALIGNÍCH MOZKOVÝCH TUMORŮ Zemanová Z. 1, Babická L. 1, Kramář F. 3, Ransdorfová Š. 2, Pavlištov tová L. 1, BřezinovB ezinová J. 2, Hrabal P. 4, Kozler P. 3, Michalová
Roman Hájek Tomáš Jelínek. Plazmocelulární leukémie (PCL)
") Roman Hájek Tomáš Jelínek Plazmocelulární leukémie (PCL) Definice (1) vzácná forma plazmocelulární dyskrázie nejagresivnější z lidských monoklonálních gamapatií incidence: 0,04/100 000 obyvatel evropské
Roman Hájek Tomáš Jelínek Plazmocelulární leukémie (PCL) Definice (1) vzácná forma plazmocelulární dyskrázie nejagresivnější z lidských monoklonálních gamapatií incidence: 0,04/100 000 obyvatel evropské
1.12.2009. Buněčné kultury. Kontinuální kultury
 Primární kultury - odvozené přímo z excise tkáně buněčné linie z různých organizmů, tkání explantované kultury jednobuněčné suspense lze je udržovat jen po omezenou dobu během kultivace ztrácejí diferenciační
Primární kultury - odvozené přímo z excise tkáně buněčné linie z různých organizmů, tkání explantované kultury jednobuněčné suspense lze je udržovat jen po omezenou dobu během kultivace ztrácejí diferenciační
NÁVAZNOST METOD KLASICKÉ A MOLEKULÁRNÍ CYTOGENETIKY. Vytvořilo Oddělení lékařské genetiky FN Brno
 NÁVAZNOST METOD KLASICKÉ A MOLEKULÁRNÍ CYTOGENETIKY TYPY CHROMOSOMOVÝCH ABERACÍ, kterých se týká vyšetření metodami klasické i molekulární cytogenetiky - VYŠETŘENÍ VROZENÝCH CHROMOSOMOVÝCH ABERACÍ prenatální
NÁVAZNOST METOD KLASICKÉ A MOLEKULÁRNÍ CYTOGENETIKY TYPY CHROMOSOMOVÝCH ABERACÍ, kterých se týká vyšetření metodami klasické i molekulární cytogenetiky - VYŠETŘENÍ VROZENÝCH CHROMOSOMOVÝCH ABERACÍ prenatální
RIGORÓZNÍ OTÁZKY - BIOLOGIE ČLOVĚKA
 RIGORÓZNÍ OTÁZKY - BIOLOGIE ČLOVĚKA 1. Genotyp a jeho variabilita, mutace a rekombinace Specifická imunitní odpověď Prevence a časná diagnostika vrozených vad 2. Genotyp a prostředí Regulace buněčného
RIGORÓZNÍ OTÁZKY - BIOLOGIE ČLOVĚKA 1. Genotyp a jeho variabilita, mutace a rekombinace Specifická imunitní odpověď Prevence a časná diagnostika vrozených vad 2. Genotyp a prostředí Regulace buněčného
Přínos molekulární genetiky pro diagnostiku a terapii malignit GIT v posledních 10 letech
 Přínos molekulární genetiky pro diagnostiku a terapii malignit GIT v posledních 10 letech Minárik M. Centrum aplikované genomiky solidních nádorů (CEGES), Genomac výzkumný ústav, Praha XXIV. JARNÍ SETKÁNÍ
Přínos molekulární genetiky pro diagnostiku a terapii malignit GIT v posledních 10 letech Minárik M. Centrum aplikované genomiky solidních nádorů (CEGES), Genomac výzkumný ústav, Praha XXIV. JARNÍ SETKÁNÍ
Pavlína Tinavská Laboratoř imunologie, Nemocnice České Budějovice
 Pavlína Tinavská Laboratoř imunologie, Nemocnice České Budějovice nízce agresivní lymfoproliferativní onemocnění základem je proliferace a akumulace klonálních maligně transformovaných vyzrálých B lymfocytů
Pavlína Tinavská Laboratoř imunologie, Nemocnice České Budějovice nízce agresivní lymfoproliferativní onemocnění základem je proliferace a akumulace klonálních maligně transformovaných vyzrálých B lymfocytů
Buněčné kultury. Kontinuální kultury
 Buněčné kultury Primární kultury - odvozené přímo z excise tkáně buněčné linie z různých organizmů, tkání explantované kultury jednobuněčné suspense lze je udržovat jen po omezenou dobu během kultivace
Buněčné kultury Primární kultury - odvozené přímo z excise tkáně buněčné linie z různých organizmů, tkání explantované kultury jednobuněčné suspense lze je udržovat jen po omezenou dobu během kultivace
ZÍSKANÉ CHROMOSOMOVÉ ABERACE. Vytvořilo Oddělení lékařské genetiky FN Brno
 ZÍSKANÉ CHROMOSOMOVÉ ABERACE CHROMOSOMOVÉ ABERACE (CHA) Cílem cytogenetického vyšetření je zjištění přítomnosti / nepřítomnosti chromosomových aberací (patologických chromosomových změn) TYPY ZÍSKANÝCH
ZÍSKANÉ CHROMOSOMOVÉ ABERACE CHROMOSOMOVÉ ABERACE (CHA) Cílem cytogenetického vyšetření je zjištění přítomnosti / nepřítomnosti chromosomových aberací (patologických chromosomových změn) TYPY ZÍSKANÝCH
Patient s hemato-onkologickým onemocněním: péče v závěru života - umírání v ČR, hospicová péče - zkušenosti jednoho pracoviště
 Patient s hemato-onkologickým onemocněním: péče v závěru života - umírání v ČR, hospicová péče - zkušenosti jednoho pracoviště Jindřich Polívka 1, Jan Švancara 2 1 Hospic Dobrého Pastýře Čerčany 2 Institut
Patient s hemato-onkologickým onemocněním: péče v závěru života - umírání v ČR, hospicová péče - zkušenosti jednoho pracoviště Jindřich Polívka 1, Jan Švancara 2 1 Hospic Dobrého Pastýře Čerčany 2 Institut
Buněčné kultury Primární kultury
 Buněčné kultury Primární kultury - odvozené přímo z excise tkáně buněčné linie z různých organizmů, tkání explantované kultury jednobuněčné suspense lze je udržovat jen po omezenou dobu během kultivace
Buněčné kultury Primární kultury - odvozené přímo z excise tkáně buněčné linie z různých organizmů, tkání explantované kultury jednobuněčné suspense lze je udržovat jen po omezenou dobu během kultivace
Co nás učí nádory? Prof. RNDr. Jana Šmardová, CSc. Ústav patologie FN Brno Přírodovědecká a Lékařská fakulta MU Brno
 Co nás učí nádory? Prof. RNDr. Jana Šmardová, CSc. Ústav patologie FN Brno Přírodovědecká a Lékařská fakulta MU Brno Brno, 17.5.2011 Izidor (Easy Door) Osnova přednášky 1. Proč nás rakovina tolik zajímá?
Co nás učí nádory? Prof. RNDr. Jana Šmardová, CSc. Ústav patologie FN Brno Přírodovědecká a Lékařská fakulta MU Brno Brno, 17.5.2011 Izidor (Easy Door) Osnova přednášky 1. Proč nás rakovina tolik zajímá?
Chromosomové změny v plasmatických buňkách u nemocných s mnohočetným myelomem detekované metodou FISH
 Chromosomové změny v plasmatických buňkách u nemocných s mnohočetným myelomem detekované metodou FISH Grant IGA NR-8183-4 J.Tajtlová, L. Pavlištová, Z. Zemanová, K. Vítovská, K. Michalová ÚKBLD CNC VFN
Chromosomové změny v plasmatických buňkách u nemocných s mnohočetným myelomem detekované metodou FISH Grant IGA NR-8183-4 J.Tajtlová, L. Pavlištová, Z. Zemanová, K. Vítovská, K. Michalová ÚKBLD CNC VFN
ZÍSKANÉ CHROMOSOMOVÉ ABERACE. Vytvořilo Oddělení lékařské genetiky FN Brno
 ZÍSKANÉ CHROMOSOMOVÉ ABERACE CHROMOSOMOVÉ ABERACE (CHA) Cílem cytogenetického vyšetření je zjištění přítomnosti / nepřítomnosti chromosomových aberací (patologických chromosomových změn) TYPY ZÍSKANÝCH
ZÍSKANÉ CHROMOSOMOVÉ ABERACE CHROMOSOMOVÉ ABERACE (CHA) Cílem cytogenetického vyšetření je zjištění přítomnosti / nepřítomnosti chromosomových aberací (patologických chromosomových změn) TYPY ZÍSKANÝCH
binding protein alpha (CEBPA) a prognostický význam mutací v jeho genu u Ota Fuchs, Arnošt t Kostečka, Monika
 a prognostický význam mutací v jeho genu u Ota Fuchs, Arnošt t Kostečka, Monika") Transkripční faktor CCAAT/enhancer binding protein alpha (CEBPA) a prognostický význam mutací v jeho genu u akutní myeloidní leukemie Ota Fuchs, Arnošt t Kostečka, Monika Holická,, Martin Vostrý, ÚHKT
Transkripční faktor CCAAT/enhancer binding protein alpha (CEBPA) a prognostický význam mutací v jeho genu u akutní myeloidní leukemie Ota Fuchs, Arnošt t Kostečka, Monika Holická,, Martin Vostrý, ÚHKT
Diagnostika a příznaky mnohočetného myelomu
 Diagnostika a příznaky mnohočetného myelomu J.Minařík, V.Ščudla Mnohočetný myelom Nekontrolované zmnožení nádorově změněných plasmatických buněk v kostní dřeni Mnohočetný = obvykle více oblastí kostní
Diagnostika a příznaky mnohočetného myelomu J.Minařík, V.Ščudla Mnohočetný myelom Nekontrolované zmnožení nádorově změněných plasmatických buněk v kostní dřeni Mnohočetný = obvykle více oblastí kostní
SEZNAM LABORATORNÍCH VYŠETŘENÍ
 Laboratoř morfologická SME 8/001/01/VERZE 01 SEZNAM LABORATORNÍCH VYŠETŘENÍ Cytologické vyšetření nátěru kostní dřeně Patologické změny krevního obrazu, klinická symptomatologie s možností hematologického
Laboratoř morfologická SME 8/001/01/VERZE 01 SEZNAM LABORATORNÍCH VYŠETŘENÍ Cytologické vyšetření nátěru kostní dřeně Patologické změny krevního obrazu, klinická symptomatologie s možností hematologického
Jak analyzovat monoklonální gamapatie
 Jak analyzovat monoklonální gamapatie -od stanovení diagnózy až po detekci léčebné odpovědi Říhová Lucie a kol. OKH, FN Brno BMG při ÚPF, LF MU Monoklonální gamapatie(mg) heterogenní skupina onemocnění
Jak analyzovat monoklonální gamapatie -od stanovení diagnózy až po detekci léčebné odpovědi Říhová Lucie a kol. OKH, FN Brno BMG při ÚPF, LF MU Monoklonální gamapatie(mg) heterogenní skupina onemocnění
Laboratoř molekulární patologie
 Laboratoř molekulární patologie Ústav patologie FN Brno Prof. RNDr. Jana Šmardová, CSc. 19.11.2014 Složení laboratoře stálí členové Prof. RNDr. Jana Šmardová, CSc. Mgr. Květa Lišková Mgr. Lenka Pitrová
Laboratoř molekulární patologie Ústav patologie FN Brno Prof. RNDr. Jana Šmardová, CSc. 19.11.2014 Složení laboratoře stálí členové Prof. RNDr. Jana Šmardová, CSc. Mgr. Květa Lišková Mgr. Lenka Pitrová
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/
 Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky Populační genetika (KBB/PG)
Inovace studia molekulární a buněčné biologie reg. č. CZ.1.07/2.2.00/07.0354 Tento projekt je spolufinancován Evropským sociálním fondem a státním rozpočtem České republiky Populační genetika (KBB/PG)
Biomarkery - diagnostika a prognóza nádorových onemocnění
 Biomarkery - diagnostika a prognóza nádorových onemocnění O. Topolčan,M.Pesta, J.Kinkorova, R. Fuchsová Fakultní nemocnice a Lékařská fakulta Plzeň CZ.1.07/2.3.00/20.0040 a IVMZČR Témata přednášky Přepdpoklady
Biomarkery - diagnostika a prognóza nádorových onemocnění O. Topolčan,M.Pesta, J.Kinkorova, R. Fuchsová Fakultní nemocnice a Lékařská fakulta Plzeň CZ.1.07/2.3.00/20.0040 a IVMZČR Témata přednášky Přepdpoklady
Vakcíny z nádorových buněk
 Protinádorové terapeutické vakcíny Vakcíny z nádorových buněk V. Vonka, ÚHKT, Praha Výhody vakcín z nádorových buněk 1.Nabízejí imunitnímu systému pacienta celé spektrum nádorových antigenů. 2. Jejich
Protinádorové terapeutické vakcíny Vakcíny z nádorových buněk V. Vonka, ÚHKT, Praha Výhody vakcín z nádorových buněk 1.Nabízejí imunitnímu systému pacienta celé spektrum nádorových antigenů. 2. Jejich
Varianty lidského chromosomu 9 z klinického i evolučního hlediska
 Varianty lidského chromosomu 9 z klinického i evolučního hlediska Antonín Šípek jr., Aleš Panczak, Romana Mihalová, Lenka Hrčková, Eva Suttrová, Mimoza Janashia a Milada Kohoutová Ústav biologie a lékařské
Varianty lidského chromosomu 9 z klinického i evolučního hlediska Antonín Šípek jr., Aleš Panczak, Romana Mihalová, Lenka Hrčková, Eva Suttrová, Mimoza Janashia a Milada Kohoutová Ústav biologie a lékařské
ÚVOD DO TRANSPLANTAČNÍ IMUNOLOGIE
 ÚVOD DO TRANSPLANTAČNÍ IMUNOLOGIE Základní funkce imunitního systému Chrání integritu organizmu proti škodlivinám zevního a vnitřního původu: chrání organizmus proti patogenním mikroorganizmům a jejich
ÚVOD DO TRANSPLANTAČNÍ IMUNOLOGIE Základní funkce imunitního systému Chrání integritu organizmu proti škodlivinám zevního a vnitřního původu: chrání organizmus proti patogenním mikroorganizmům a jejich
VLIV ČASNÉ BASOFILIE NA PROGNÓZU CML. Autoři: Lucie Benešová, Barbora Maiwaldová. Výskyt
 VLIV ČASNÉ BASOFILIE NA PROGNÓZU CML Autoři: Lucie Benešová, Barbora Maiwaldová Výskyt Chronická myeloidní leukémie je nejčastěji se vyskytujícím myeloproliferativním onemocněním. Obvykle se udává, že
VLIV ČASNÉ BASOFILIE NA PROGNÓZU CML Autoři: Lucie Benešová, Barbora Maiwaldová Výskyt Chronická myeloidní leukémie je nejčastěji se vyskytujícím myeloproliferativním onemocněním. Obvykle se udává, že
Lékařská genetika a onkologie. Renata Gaillyová OLG a LF MU Brno 2012/2013
 Lékařská genetika a onkologie Renata Gaillyová OLG a LF MU Brno 2012/2013 *genetické souvislosti *onkogenetická vyšetření u onkologických onemocnění * genetické vyšetření u hereditárních nádorů *presymptomatické
Lékařská genetika a onkologie Renata Gaillyová OLG a LF MU Brno 2012/2013 *genetické souvislosti *onkogenetická vyšetření u onkologických onemocnění * genetické vyšetření u hereditárních nádorů *presymptomatické
 KLASICKÉ A MOLEKULÁRN RNÍ METODY V KLINICKÉ A ONKOLOGICKÉ CYTOGENETICE. Zuzana Zemanová CENTRUM NÁDOROVN DOROVÉ CYTOGENETIKY Ústav klinické biochemie a laboratorní diagnostiky VFN a 1. LF UK PRAHA CYTOGENETIKA
KLASICKÉ A MOLEKULÁRN RNÍ METODY V KLINICKÉ A ONKOLOGICKÉ CYTOGENETICE. Zuzana Zemanová CENTRUM NÁDOROVN DOROVÉ CYTOGENETIKY Ústav klinické biochemie a laboratorní diagnostiky VFN a 1. LF UK PRAHA CYTOGENETIKA
NÁLEZ DVOJITĚ POZITIVNÍCH T LYMFOCYTŮ - CO TO MŮŽE ZNAMENAT? Ondřej Souček Ústav klinické imunologie a alergologie Fakultní nemocnice Hradec Králové
 NÁLEZ DVOJITĚ POZITIVNÍCH T LYMFOCYTŮ - CO TO MŮŽE ZNAMENAT? Ondřej Souček Ústav klinické imunologie a alergologie Fakultní nemocnice Hradec Králové LEUKÉMIE x LYMFOM Nádorová onemocnění buněk krvetvorné
NÁLEZ DVOJITĚ POZITIVNÍCH T LYMFOCYTŮ - CO TO MŮŽE ZNAMENAT? Ondřej Souček Ústav klinické imunologie a alergologie Fakultní nemocnice Hradec Králové LEUKÉMIE x LYMFOM Nádorová onemocnění buněk krvetvorné
Bioptická laboratoř s.r.o. a Šiklův ústav patologie Lékařské fakulty UK v Plzni
 Genové fúze NCOA4-RET a TRIM27- RETdiferencují intraduktální karcinom slinných žláz na duktální a apokrinní podtyp: analýza 18 případů pomocí sekvenování nové generace (NGS) Skálová A, Baněčková M, Martínek
Genové fúze NCOA4-RET a TRIM27- RETdiferencují intraduktální karcinom slinných žláz na duktální a apokrinní podtyp: analýza 18 případů pomocí sekvenování nové generace (NGS) Skálová A, Baněčková M, Martínek
Adobe Captivate Wednesday, January 09, 2013. Slide 1 - Zhoubné nádory u dětí epidemiologie, odlišnosti od nádorů dospělých, nádorové markery
 Slide 1 - Zhoubné nádory u dětí epidemiologie, odlišnosti od nádorů dospělých, nádorové markery Page 1 of 45 Slide 2 - Zastoupení nádorů u dětí Page 2 of 45 Slide 3 - Epidemiologie nádorů dětského věku
Slide 1 - Zhoubné nádory u dětí epidemiologie, odlišnosti od nádorů dospělých, nádorové markery Page 1 of 45 Slide 2 - Zastoupení nádorů u dětí Page 2 of 45 Slide 3 - Epidemiologie nádorů dětského věku
Leukémie. - onemocnění postihující hemopoetický systém. vznik hromaděním změn v genomu kmenových buněk progenitorů jednotlivých řad
 Nádory dětskd tského věkuv 30-35% 35% leukémie 11% lymfomy 28% nádory n CNS Neuroblastom 6-8% nefroblastom 6% rabdomyosarkom 3% osteosarkom Retinoblastom 2% EWS Hepatoblastom Germináln lní nádory Leukémie
Nádory dětskd tského věkuv 30-35% 35% leukémie 11% lymfomy 28% nádory n CNS Neuroblastom 6-8% nefroblastom 6% rabdomyosarkom 3% osteosarkom Retinoblastom 2% EWS Hepatoblastom Germináln lní nádory Leukémie
Tekuté biopsie u mnohočetného myelomu
 Tekuté biopsie u mnohočetného myelomu Mgr. Veronika Kubaczková Babákova myelomová skupina ÚPF LF MU Pacientský seminář 11. května 2016, Brno Co jsou tekuté biopsie? Představují méně zatěžující vyšetření
Tekuté biopsie u mnohočetného myelomu Mgr. Veronika Kubaczková Babákova myelomová skupina ÚPF LF MU Pacientský seminář 11. května 2016, Brno Co jsou tekuté biopsie? Představují méně zatěžující vyšetření
MYELOFIBROSA - DIAGNOSTIKA A LÉČEBNÉ MOŽNOSTI
 MYELOFIBROSA - DIAGNOSTIKA A LÉČEBNÉ MOŽNOSTI DEFINICE: chronické myeloproliferativní (klonální) onemocnění charakteristické zmnožením vaziva v kostní dřeni a extramedulární krvetvorbou (první popis -
MYELOFIBROSA - DIAGNOSTIKA A LÉČEBNÉ MOŽNOSTI DEFINICE: chronické myeloproliferativní (klonální) onemocnění charakteristické zmnožením vaziva v kostní dřeni a extramedulární krvetvorbou (první popis -
Diagnostika genetických změn u papilárního karcinomu štítné žlázy
 Diagnostika genetických změn u papilárního karcinomu štítné žlázy Vlasta Sýkorová Oddělení molekulární endokrinologie Endokrinologický ústav, Praha Nádory štítné žlázy folikulární buňka parafolikulární
Diagnostika genetických změn u papilárního karcinomu štítné žlázy Vlasta Sýkorová Oddělení molekulární endokrinologie Endokrinologický ústav, Praha Nádory štítné žlázy folikulární buňka parafolikulární
Příloha I. Vědecké závěry a zdůvodnění změny v registraci
 Příloha I Vědecké závěry a zdůvodnění změny v registraci 1 Vědecké závěry S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizované zprávy/aktualizovaných zpráv o bezpečnosti (PSUR)
Příloha I Vědecké závěry a zdůvodnění změny v registraci 1 Vědecké závěry S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizované zprávy/aktualizovaných zpráv o bezpečnosti (PSUR)
Chromozomální aberace nalezené u párů s poruchou reprodukce v letech
 Chromozomální aberace nalezené u párů s poruchou reprodukce v letech 2000-2005 Jak přistupovat k nálezům minoritních gonozomálních mozaik? Šantavá A., Adamová, K.,Čapková P., Hyjánek J. Ústav lékařské
Chromozomální aberace nalezené u párů s poruchou reprodukce v letech 2000-2005 Jak přistupovat k nálezům minoritních gonozomálních mozaik? Šantavá A., Adamová, K.,Čapková P., Hyjánek J. Ústav lékařské
Proměnlivost organismu. Mgr. Aleš RUDA
 Proměnlivost organismu Mgr. Aleš RUDA Faktory variability organismů Vnitřní = faktory vedoucí k proměnlivosti genotypu Vnější = faktory prostředí Příčiny proměnlivosti děje probíhající při meioze segregace
Proměnlivost organismu Mgr. Aleš RUDA Faktory variability organismů Vnitřní = faktory vedoucí k proměnlivosti genotypu Vnější = faktory prostředí Příčiny proměnlivosti děje probíhající při meioze segregace
KLONÁLN LNÍ CHROMOSOMOVÉ ABERACE U MNOHOČETN ETNÉHO MYELOMU (MM). Klasické G-pruhovací techniky patologický nález n aža 40% nemocných Cca 60% případp
. Klasické G-pruhovací techniky patologický nález n aža 40% nemocných Cca 60% případp") CHROMOSOMOVÉ ABERACE U MNOHOČETN ETNÉHO MYELOMU - Molekulárn rně cytogenetická analýza plasmatických buněk Zuzana Zemanová 1, Lenka Pavlištov tová 1, Jana Tajtlová 1, Ivan Špička 2, Evžen Gregora 3, Kyra
CHROMOSOMOVÉ ABERACE U MNOHOČETN ETNÉHO MYELOMU - Molekulárn rně cytogenetická analýza plasmatických buněk Zuzana Zemanová 1, Lenka Pavlištov tová 1, Jana Tajtlová 1, Ivan Špička 2, Evžen Gregora 3, Kyra
Potřebné genetické testy pro výzkum a jejich dostupnost, spolupráce s neurology Taťána Maříková. Parent projekt. Praha 19.2.2009
 Potřebné genetické testy pro výzkum a jejich dostupnost, spolupráce s neurology Taťána Maříková Parent projekt Praha 19.2.2009 Diagnostika MD její vývoj 1981-1986: zdokonalování diferenciální diagnostiky
Potřebné genetické testy pro výzkum a jejich dostupnost, spolupráce s neurology Taťána Maříková Parent projekt Praha 19.2.2009 Diagnostika MD její vývoj 1981-1986: zdokonalování diferenciální diagnostiky
7. Regulace genové exprese, diferenciace buněk a epigenetika
 7. Regulace genové exprese, diferenciace buněk a epigenetika Aby mohl mnohobuněčný organismus efektivně fungovat, je třeba, aby se jednotlivé buňky specializovaly na určité funkce. Nový jedinec přitom
7. Regulace genové exprese, diferenciace buněk a epigenetika Aby mohl mnohobuněčný organismus efektivně fungovat, je třeba, aby se jednotlivé buňky specializovaly na určité funkce. Nový jedinec přitom
FUNKČNÍ VARIANTA GENU ANXA11 SNIŽUJE RIZIKO ONEMOCNĚNÍ
 FUNKČNÍ VARIANTA GENU ANXA11 SNIŽUJE RIZIKO ONEMOCNĚNÍ SARKOIDÓZOU: POTVRZENÍ VÝSLEDKŮ CELOGENOMOVÉ ASOCIAČNÍ STUDIE. Sťahelová A. 1, Mrázek F. 1, Kriegová E. 1, Hutyrová B. 2, Kubištová Z. 1, Kolek V.
FUNKČNÍ VARIANTA GENU ANXA11 SNIŽUJE RIZIKO ONEMOCNĚNÍ SARKOIDÓZOU: POTVRZENÍ VÝSLEDKŮ CELOGENOMOVÉ ASOCIAČNÍ STUDIE. Sťahelová A. 1, Mrázek F. 1, Kriegová E. 1, Hutyrová B. 2, Kubištová Z. 1, Kolek V.
Přehled výzkumných aktivit
 Přehled výzkumných aktivit ROK 2004 Lenka Zahradová Laboratoř experimentální hematologie a buněčné imunoterapie Oddělení klinické hematologie FNB Bohunice Přednosta: prof. MUDr. M. Penka, CSc. Oddělení
Přehled výzkumných aktivit ROK 2004 Lenka Zahradová Laboratoř experimentální hematologie a buněčné imunoterapie Oddělení klinické hematologie FNB Bohunice Přednosta: prof. MUDr. M. Penka, CSc. Oddělení
Základy molekulární biologie KBC/MBIOZ
 Základy molekulární biologie KBC/MBIOZ Mária Čudejková 2. Transkripce genu a její regulace Transkripce genetické informace z DNA na RNA Transkripce dvou genů zachycená na snímku z elektronového mikroskopu.
Základy molekulární biologie KBC/MBIOZ Mária Čudejková 2. Transkripce genu a její regulace Transkripce genetické informace z DNA na RNA Transkripce dvou genů zachycená na snímku z elektronového mikroskopu.
Příčiny a projevy abnormálního vývoje
 Příčiny a projevy abnormálního vývoje Ústav histologie a embryologie 1. LF UK v Praze MUDr. Filip Wagner Předmět: Obecná histologie a obecná embryologie (B02241) 1 Vrozené vývojové vady vývojové poruchy
Příčiny a projevy abnormálního vývoje Ústav histologie a embryologie 1. LF UK v Praze MUDr. Filip Wagner Předmět: Obecná histologie a obecná embryologie (B02241) 1 Vrozené vývojové vady vývojové poruchy
Epidemiologie hematologických malignit v České republice
 Epidemiologie hematologických malignit v České republice Zpráva je plněním Národního onkologického programu ČR a vychází z uzavřených a validovaných dat Národního onkologického registru za období 1976
Epidemiologie hematologických malignit v České republice Zpráva je plněním Národního onkologického programu ČR a vychází z uzavřených a validovaných dat Národního onkologického registru za období 1976
Odběr krvetvorných buněk z periferní krve: příprava, průběh a komplikace
 Odběr krvetvorných buněk z periferní krve: příprava, průběh a komplikace Helena Švábová, Andrea Žmijáková Interní hematologická a onkologická klinika FN Brno Separační středisko je součástí Interní hematologické
Odběr krvetvorných buněk z periferní krve: příprava, průběh a komplikace Helena Švábová, Andrea Žmijáková Interní hematologická a onkologická klinika FN Brno Separační středisko je součástí Interní hematologické
Prof. MUDr. Jiří Vorlíček, CSc. Prof. MUDr. Jitka Abrahámová, DrSc. MUDr. Tomáš Büchler, PhD.
 Promítnutí pokroků lékařské vědy do funkčního hodnocení zdravotního stavu a pracovní schopnosti ve vztahu k mezinárodní klasifikaci nemocí a s přihlédnutím k Mezinárodní klasifikaci funkčních schopností
Promítnutí pokroků lékařské vědy do funkčního hodnocení zdravotního stavu a pracovní schopnosti ve vztahu k mezinárodní klasifikaci nemocí a s přihlédnutím k Mezinárodní klasifikaci funkčních schopností
Mimodřeňová expanze plazmocytů do CNS u mnohočetného myelomu
 Setkání uživatelů Průtokové cytometrie Beckman Coulter, 13.-14.5.2018, Valeč Mimodřeňová expanze plazmocytů do CNS u mnohočetného myelomu Říhová Lucie a kol. OKH, FN Brno BMG při ÚPF, LF MU Lokalizace
Setkání uživatelů Průtokové cytometrie Beckman Coulter, 13.-14.5.2018, Valeč Mimodřeňová expanze plazmocytů do CNS u mnohočetného myelomu Říhová Lucie a kol. OKH, FN Brno BMG při ÚPF, LF MU Lokalizace
Projekt MGUS V.Sandecká, R.Hájek, J.Radocha, V.Maisnar. Velké Bílovice
 Projekt MGUS 2010 V.Sandecká, R.Hájek, J.Radocha, V.Maisnar CZECH CMG ČESKÁ MYELOMOVÁ SKUPINA M Y E L O M A NADAČNÍ FOND GROUP Velké Bílovice 25.4.2009 Česká myelomová skupina a její nadační fond spolupracují
Projekt MGUS 2010 V.Sandecká, R.Hájek, J.Radocha, V.Maisnar CZECH CMG ČESKÁ MYELOMOVÁ SKUPINA M Y E L O M A NADAČNÍ FOND GROUP Velké Bílovice 25.4.2009 Česká myelomová skupina a její nadační fond spolupracují
http://www.accessexcellence.org/ab/gg/chromosome.html
 3. cvičení Buněčný cyklus Mitóza Modifikace mitózy 1 DNA, chromosom genetická informace organismu chromosom = strukturní podoba DNA během dělení (mitózy) řetězec DNA (chromonema) histony další enzymatické
3. cvičení Buněčný cyklus Mitóza Modifikace mitózy 1 DNA, chromosom genetická informace organismu chromosom = strukturní podoba DNA během dělení (mitózy) řetězec DNA (chromonema) histony další enzymatické
Sekreční karcinom slinných žláz: využití genomového profilování v personalizaci onkologické léčby: analýza 49 případů sekvenováním nové generace (NGS)
") Sekreční karcinom slinných žláz: využití genomového profilování v personalizaci onkologické léčby: analýza 49 případů sekvenováním nové generace (NGS) Skálová A, Santana T, Baněčková M, Vaněček T, Michal
Sekreční karcinom slinných žláz: využití genomového profilování v personalizaci onkologické léčby: analýza 49 případů sekvenováním nové generace (NGS) Skálová A, Santana T, Baněčková M, Vaněček T, Michal
EPIGENETIKA reverzibilních změn funkce genů, Epigenetické faktory ovlivňují fenotyp bez změny genotypu. Epigenetická
 EPIGENETIKA Epigenetika se zabývá studiem reverzibilních změn funkce genů, aniž by při tom došlo ke změnám v sekvenci jaderné DNA. Epigenetické faktory ovlivňují fenotyp bez změny genotypu. Epigenetická
EPIGENETIKA Epigenetika se zabývá studiem reverzibilních změn funkce genů, aniž by při tom došlo ke změnám v sekvenci jaderné DNA. Epigenetické faktory ovlivňují fenotyp bez změny genotypu. Epigenetická
Základy klinické cytogenetiky chromosomy
 Základy klinické cytogenetiky chromosomy Hanáková M. SHRNUTÍ PŘEDNÁŠKY chromosomy metody přípravy chromosomových preparátů, hodnocení chromosomů, metody molekulární cytogenetiky vrozené chromosomové aberace
Základy klinické cytogenetiky chromosomy Hanáková M. SHRNUTÍ PŘEDNÁŠKY chromosomy metody přípravy chromosomových preparátů, hodnocení chromosomů, metody molekulární cytogenetiky vrozené chromosomové aberace
Glosář - Cestina. Odchylka počtu chromozomů v jádře buňky od normy. Např. 45 nebo 47 chromozomů místo obvyklých 46. Příkladem je trizomie 21
 Glosář - Cestina alely aneuploidie asistovaná reprodukce autozomálně dominantní autozomálně recesivní BRCA chromozom chromozomová aberace cytogenetický laborant de novo Různé formy genu, které se nacházejí
Glosář - Cestina alely aneuploidie asistovaná reprodukce autozomálně dominantní autozomálně recesivní BRCA chromozom chromozomová aberace cytogenetický laborant de novo Různé formy genu, které se nacházejí
Projekt CAMELIA Projekt ALERT
 Projekt Alert akutní leukemie klinický registr x Web projektu Diskusní klub projektu Management dat Služby IS Help Zpět Analytické nástroje Prohlížeč dat Expertní služby x Projekt CAMELIA Projekt ALERT
Projekt Alert akutní leukemie klinický registr x Web projektu Diskusní klub projektu Management dat Služby IS Help Zpět Analytické nástroje Prohlížeč dat Expertní služby x Projekt CAMELIA Projekt ALERT
Návrh směrnic pro správnou laboratorní diagnostiku Friedreichovy ataxie.
 Návrh směrnic pro správnou laboratorní diagnostiku Friedreichovy ataxie. Připravila L.Fajkusová Online Mendelian Inheritance in Man: #229300 FRIEDREICH ATAXIA 1; FRDA *606829 FRDA GENE; FRDA Popis onemocnění
Návrh směrnic pro správnou laboratorní diagnostiku Friedreichovy ataxie. Připravila L.Fajkusová Online Mendelian Inheritance in Man: #229300 FRIEDREICH ATAXIA 1; FRDA *606829 FRDA GENE; FRDA Popis onemocnění
Downův syndrom. Renata Gaillyová OLG FN Brno
 Downův syndrom Renata Gaillyová OLG FN Brno Zastoupení genetických chorob a vývojových vad podle etiologie 0,6 %-0,7% populace má vrozenou chromosomovou aberaci incidence vážných monogenně podmíněných
Downův syndrom Renata Gaillyová OLG FN Brno Zastoupení genetických chorob a vývojových vad podle etiologie 0,6 %-0,7% populace má vrozenou chromosomovou aberaci incidence vážných monogenně podmíněných
CEBO: (Center for Evidence Based Oncology) Incidence Kostních příhod u nádorů prsu PROJEKT IKARUS. Neintervenční epidemiologická studie
 Incidence Kostních příhod u nádorů prsu PROJEKT IKARUS. Neintervenční epidemiologická studie") CEBO: (Center for Evidence Based Oncology) Incidence Kostních příhod u nádorů prsu PROJEKT Neintervenční epidemiologická studie PROTOKOL PROJEKTU Verze: 4.0 Datum: 26.09.2006 Strana 2 PROTOKOL PROJEKTU
CEBO: (Center for Evidence Based Oncology) Incidence Kostních příhod u nádorů prsu PROJEKT Neintervenční epidemiologická studie PROTOKOL PROJEKTU Verze: 4.0 Datum: 26.09.2006 Strana 2 PROTOKOL PROJEKTU
Buněčný cyklus. Replikace DNA a dělení buňky
 Buněčný cyklus Replikace DNA a dělení buňky 2 Regulace buněčného dělení buněčný cyklus: buněčné dělení buněčný růst kontrola kvality potomstva (dceřinných buněk) bránípřenosu nekompletně zreplikovaných
Buněčný cyklus Replikace DNA a dělení buňky 2 Regulace buněčného dělení buněčný cyklus: buněčné dělení buněčný růst kontrola kvality potomstva (dceřinných buněk) bránípřenosu nekompletně zreplikovaných
Urychlení úpravy krvetvorby poškozené cytostatickou terapií (5-fluorouracil a cisplatina) p.o. aplikací IMUNORu
 p.o. aplikací IMUNORu") Urychlení úpravy krvetvorby poškozené cytostatickou terapií (5-fluorouracil a cisplatina) p.o. aplikací IMUNORu Úvod Myelosuprese (poškození krvetvorby) patří mezi nejčastější vedlejší účinky chemoterapie.
Urychlení úpravy krvetvorby poškozené cytostatickou terapií (5-fluorouracil a cisplatina) p.o. aplikací IMUNORu Úvod Myelosuprese (poškození krvetvorby) patří mezi nejčastější vedlejší účinky chemoterapie.
Genetický polymorfismus
 Genetický polymorfismus Za geneticky polymorfní je považován znak s nejméně dvěma geneticky podmíněnými variantami v jedné populaci, které se nachází v takových frekvencích, že i zřídkavá má frekvenci
Genetický polymorfismus Za geneticky polymorfní je považován znak s nejméně dvěma geneticky podmíněnými variantami v jedné populaci, které se nachází v takových frekvencích, že i zřídkavá má frekvenci
Časná a pozdní toxicita léčby lymfomů. David Belada FN a LF UK v Hradci Králové
 Časná a pozdní toxicita léčby lymfomů David Belada FN a LF UK v Hradci Králové Co je to toxicita léčby? Toxicita léčby lymfomů Jaký je rozdíl mezi časnou a pozdní toxicitou? Dá se toxicita předvídat? Existuje
Časná a pozdní toxicita léčby lymfomů David Belada FN a LF UK v Hradci Králové Co je to toxicita léčby? Toxicita léčby lymfomů Jaký je rozdíl mezi časnou a pozdní toxicitou? Dá se toxicita předvídat? Existuje
Zuzana Zemanová 1, Filip Kramář 2
 CHROMOSOMOVÉ ABERACE V BUŇKÁCH MALIGNÍCH MOZKOVÝCH NÁDORŮ. Zuzana Zemanová 1, Filip Kramář 2 1 Centrum nádorové cytogenetiky, Ústav klinické biochemie a laboratorní diagnostiky, Všeobecná fakultní nemocnice
CHROMOSOMOVÉ ABERACE V BUŇKÁCH MALIGNÍCH MOZKOVÝCH NÁDORŮ. Zuzana Zemanová 1, Filip Kramář 2 1 Centrum nádorové cytogenetiky, Ústav klinické biochemie a laboratorní diagnostiky, Všeobecná fakultní nemocnice
Rozdíly mezi chronickou myeloidní leukemií (CML) a chronickou lymfocytární leukemií (CLL)
 a chronickou lymfocytární leukemií (CLL)") Rozdíly mezi chronickou myeloidní leukemií (CML) a chronickou lymfocytární leukemií (CLL) Stanislava Hrobková, Michael Doubek Interní hematologická a onkologická klinika LF MU a FN Brno Co je to leukemie
Rozdíly mezi chronickou myeloidní leukemií (CML) a chronickou lymfocytární leukemií (CLL) Stanislava Hrobková, Michael Doubek Interní hematologická a onkologická klinika LF MU a FN Brno Co je to leukemie
Variabilita heterochromatinové oblasti lidského chromosomu 9 z evolučního a klinického hlediska
 Variabilita heterochromatinové oblasti lidského chromosomu 9 z evolučního a klinického hlediska A. Šípek jr. (1), R. Mihalová (1), A. Panczak (1), L. Hrčková (1), P. Lonský (2), M. Janashia (1), M. Kohoutová
Variabilita heterochromatinové oblasti lidského chromosomu 9 z evolučního a klinického hlediska A. Šípek jr. (1), R. Mihalová (1), A. Panczak (1), L. Hrčková (1), P. Lonský (2), M. Janashia (1), M. Kohoutová
LÉKAŘSKÁ VYŠETŘENÍ A LABORATORNÍ TESTY
 LÉKAŘSKÁ VYŠETŘENÍ A LABORATORNÍ TESTY Pokud čtete tento text, pravděpodobně jste v kontaktu s odborníkem na léčbu mnohočetného myelomu. Diagnóza mnohočetného myelomu je stanovena pomocí četných laboratorních
LÉKAŘSKÁ VYŠETŘENÍ A LABORATORNÍ TESTY Pokud čtete tento text, pravděpodobně jste v kontaktu s odborníkem na léčbu mnohočetného myelomu. Diagnóza mnohočetného myelomu je stanovena pomocí četných laboratorních
Genetická kontrola prenatáln. lního vývoje
 Genetická kontrola prenatáln lního vývoje Stádia prenatáln lního vývoje Preembryonální stádium do 6. dne po oplození zygota až blastocysta polární organizace cytoplasmatických struktur zygoty Embryonální
Genetická kontrola prenatáln lního vývoje Stádia prenatáln lního vývoje Preembryonální stádium do 6. dne po oplození zygota až blastocysta polární organizace cytoplasmatických struktur zygoty Embryonální
Studium genetické predispozice ke vzniku karcinomu prsu
 Univerzita Karlova v Praze 1. lékařská fakulta Studium genetické predispozice ke vzniku karcinomu prsu Petra Kleiblová Ústav biochemie a experimentální onkologie, 1. LF UK - skupina molekulární biologie
Univerzita Karlova v Praze 1. lékařská fakulta Studium genetické predispozice ke vzniku karcinomu prsu Petra Kleiblová Ústav biochemie a experimentální onkologie, 1. LF UK - skupina molekulární biologie
Mutace genu pro Connexin 26 jako významná příčina nedoslýchavosti
 Mutace genu pro Connexin 26 jako významná příčina nedoslýchavosti Petr Lesný 1, Pavel Seeman 2, Daniel Groh 1 1 ORL klinika UK 2. LF a FN Motol Subkatedra dětské ORL IPVZ Přednosta doc. MUDr. Zdeněk Kabelka
Mutace genu pro Connexin 26 jako významná příčina nedoslýchavosti Petr Lesný 1, Pavel Seeman 2, Daniel Groh 1 1 ORL klinika UK 2. LF a FN Motol Subkatedra dětské ORL IPVZ Přednosta doc. MUDr. Zdeněk Kabelka
VĚDA A VÝZKUM V PERIOPERAČNÍ PÉČI. Mgr. Markéta Jašková Dana Svobodová Gynekologicko-porodnická klinika Fakultní nemocnice Ostrava
 VĚDA A VÝZKUM V PERIOPERAČNÍ PÉČI Mgr. Markéta Jašková Dana Svobodová Gynekologicko-porodnická klinika Fakultní nemocnice Ostrava VĚDA A VÝZKUM NA GOS Detekce mutace genu BRCA1 a BRCA2, a to přímo z nádorové
VĚDA A VÝZKUM V PERIOPERAČNÍ PÉČI Mgr. Markéta Jašková Dana Svobodová Gynekologicko-porodnická klinika Fakultní nemocnice Ostrava VĚDA A VÝZKUM NA GOS Detekce mutace genu BRCA1 a BRCA2, a to přímo z nádorové
Co je to transplantace krvetvorných buněk?
 Co je to transplantace krvetvorných buněk? Transplantace krvetvorných buněk je přenos vlastní (autologní) nebo dárcovské (alogenní) krvetvorné tkáně. Účelem je obnova kostní dřeně po vysoce dávkové chemoterapii
Co je to transplantace krvetvorných buněk? Transplantace krvetvorných buněk je přenos vlastní (autologní) nebo dárcovské (alogenní) krvetvorné tkáně. Účelem je obnova kostní dřeně po vysoce dávkové chemoterapii
Vliv věku rodičů při početí na zdraví dítěte
 Vliv věku rodičů při početí na zdraví dítěte Antonín Šípek Jr 1,2, Vladimír Gregor 2,3, Antonín Šípek 2,3,4 1) Ústav biologie a lékařské genetiky 1. LF UK a VFN, Praha 2) Oddělení lékařské genetiky, Thomayerova
Vliv věku rodičů při početí na zdraví dítěte Antonín Šípek Jr 1,2, Vladimír Gregor 2,3, Antonín Šípek 2,3,4 1) Ústav biologie a lékařské genetiky 1. LF UK a VFN, Praha 2) Oddělení lékařské genetiky, Thomayerova
1. Definice a historie oboru molekulární medicína. 3. Základní laboratorní techniky v molekulární medicíně
 Obsah Předmluvy 1. Definice a historie oboru molekulární medicína 1.1. Historie molekulární medicíny 2. Základní principy molekulární biologie 2.1. Historie molekulární biologie 2.2. DNA a chromozomy 2.3.
Obsah Předmluvy 1. Definice a historie oboru molekulární medicína 1.1. Historie molekulární medicíny 2. Základní principy molekulární biologie 2.1. Historie molekulární biologie 2.2. DNA a chromozomy 2.3.
JEDINEČNÁ INFORMACE. Jediný prenatální krevní test, který analyzuje všechny chromozomy vašeho miminka
 JEDINEČNÁ INFORMACE Jediný prenatální krevní test, který analyzuje všechny chromozomy vašeho miminka MaterniT GENOME test nabízí více informací o chromozomech vašeho miminka než kterýkoliv jiný prenatální
JEDINEČNÁ INFORMACE Jediný prenatální krevní test, který analyzuje všechny chromozomy vašeho miminka MaterniT GENOME test nabízí více informací o chromozomech vašeho miminka než kterýkoliv jiný prenatální
Výuka genetiky na PřF OU K. MALACHOVÁ
 Výuka genetiky na PřF OU K. MALACHOVÁ KATEDRA BIOLOGIE A EKOLOGIE BAKALÁŘSKÉ STUDIJNÍ PROGRAMY Experimentální Systematická Aplikovaná (prezenční, kombinovaná) Jednooborová Dvouoborová KATEDRA BIOLOGIE
Výuka genetiky na PřF OU K. MALACHOVÁ KATEDRA BIOLOGIE A EKOLOGIE BAKALÁŘSKÉ STUDIJNÍ PROGRAMY Experimentální Systematická Aplikovaná (prezenční, kombinovaná) Jednooborová Dvouoborová KATEDRA BIOLOGIE
Skrytá hrozba - jaké chromosomové aberace nezachytí prosté vyloučení nejčastějších aneuploidií?
 Skrytá hrozba - jaké chromosomové aberace nezachytí prosté vyloučení nejčastějších aneuploidií? Antonín Šípek Jr 1,2, Vladimír Gregor 2,3, Antonín Šípek 2,3,4 1. Ústav biologie a lékařské genetiky 1. LF
Skrytá hrozba - jaké chromosomové aberace nezachytí prosté vyloučení nejčastějších aneuploidií? Antonín Šípek Jr 1,2, Vladimír Gregor 2,3, Antonín Šípek 2,3,4 1. Ústav biologie a lékařské genetiky 1. LF
Zkušební otázky z oboru hematologie 2. ročník bakalářského studia LF MU obor Zdravotní laborant
 Zkušební otázky z oboru hematologie 2. ročník bakalářského studia LF MU obor Zdravotní laborant I. Praktická zkouška z laboratorní hematologie IA) Část morfologická 1. Hematopoéza - vývojové krevní řady
Zkušební otázky z oboru hematologie 2. ročník bakalářského studia LF MU obor Zdravotní laborant I. Praktická zkouška z laboratorní hematologie IA) Část morfologická 1. Hematopoéza - vývojové krevní řady
Mgr. Zuzana Kufová , Mikulov. Genomické analýzy u Waldenströmovy makroglobulinémie
 Mgr. Zuzana Kufová 10.4.2015, Mikulov Genomické analýzy u Waldenströmovy makroglobulinémie Obsah přednášky Genomický profil u WM Mutace v genu MYD88 Mutace v genu CXCR4 Diagnostika Výsledky http://socratic.org/questions/how-do-functional-groups-affect-the-structure-and-function-of-macromolecules
Mgr. Zuzana Kufová 10.4.2015, Mikulov Genomické analýzy u Waldenströmovy makroglobulinémie Obsah přednášky Genomický profil u WM Mutace v genu MYD88 Mutace v genu CXCR4 Diagnostika Výsledky http://socratic.org/questions/how-do-functional-groups-affect-the-structure-and-function-of-macromolecules
Patologie krevního ústrojí. II. histologické praktikum 3. ročník všeobecného směru
 Patologie krevního ústrojí II. histologické praktikum 3. ročník všeobecného směru H&E Polycythaemia vera Giemsa Erythroidní proliferace Glycophorin C Polycythaemia vera ABNORMAL MEGAKARYOCYTIC PROLIFERATION
Patologie krevního ústrojí II. histologické praktikum 3. ročník všeobecného směru H&E Polycythaemia vera Giemsa Erythroidní proliferace Glycophorin C Polycythaemia vera ABNORMAL MEGAKARYOCYTIC PROLIFERATION
Molekulárně cytogenetické vyšetření pacientů s mnohočetným myelomem na OLG FN Brno
 Molekulárně cytogenetické vyšetření pacientů s mnohočetným myelomem na OLG FN Brno Filková H., Oltová A., Kuglík P.Vranová V., Kupská R., Strašilová J., Hájek R. OLG FN Brno, PřF MU Brno, IHOK FN Brno
Molekulárně cytogenetické vyšetření pacientů s mnohočetným myelomem na OLG FN Brno Filková H., Oltová A., Kuglík P.Vranová V., Kupská R., Strašilová J., Hájek R. OLG FN Brno, PřF MU Brno, IHOK FN Brno